


UNEARTHING OF NATURAL BIOACTIVES: AN OVERVIEW OF COMPUTATIONAL APPROACHES
HTML Full TextUNEARTHING OF NATURAL BIOACTIVES: AN OVERVIEW OF COMPUTATIONAL APPROACHES
Simran Chaurasia
Department of Pharmaceutical Sciences and Technology, Maharaja Ranjit Singh Punjab Technical University, Badal Road, Bathinda, Punjab, India.
ABSTRACT: The computer-aided drug design (CADD) symbolizes an intricate discipline exploitation accomplishment in varied areas of science and varied strategies and approaches. It tends to speed-up and optimize discovery of latest biologically active compounds. However, these approaches cannot replace the experimental tests. The aim of CADD is to generate hypotheses regarding probable new ligands and their interaction with targets. It's considered that these strategies will decrease the quantity of the compounds that are required to be synthesized and tested for biological activity up to 2 orders. Thus, they're capable to decrease basically time consuming monetary expenses for development of medicine. A Recent terribly effective technique in era is drug designing through computers. CADD technologies are employed in technology, biological science, organic chemistry etc. The main advantage of the CADD is value effective in analysis and development of medicine. There are wide ranges of software that are employed in CADD, Grid computing, window primarily based general PBPK/PD modeling computer software, PKUDDS for structure based drug design, APIS, JAVA, Perl and Python. The various techniques that are enclosed in CADD are visualization, homology, molecular dynamic, energy minimization molecular docking, QSAR etc. CADD is applicable in cancer disease, transportation of drug to specific site in body, information collections and storages of organics and biologicals. This article conjointly deals with the development of structure-based drug design for human herpes virus (HHV) infection that implicates selecting the target proteins, visualizing the target structure, identifying the binding site, docking up the ligands and evaluating those using computational techniques.
Keywords: Averrhoa carambola Leaves, Pharmacognostical, Phytochemical
INTRODUCTION: Drug discovery and development is time taking, and resource consuming process which is full of risk. Numerous new technologies have been developed and applied in drug Research & Development to shorten their search cycle and to reduce the expenses 1. Computational approaches have revolutionized the pipeline of discovery and development.
Last four decades are witness of rapid advancement in computational technology in drug research and development (R&D) including computer applications in the development of biomedicines. In the post genomic era, computational tools have been applied in almost every stage of research due to enormous increase in the information regarding the biomolecules, which ultimately affect the strategies for drug discovery.
It helps in discovery and development of pipeline, from target identification to lead discovery, from lead optimization to preclinical or clinical trials. The advances of computational technologies for drug discovery and development; emphases are put on computational tools for identification of target, discovery of lead molecule, as well as pharmacokinetic parameters including absorption, distribution, metabolism, excretion, and toxicity prediction. The concept depicted in Fig. 1 starts with computational tool for getting an idea of putative hits from nature. As soon as a sensitive data-mining tool has been developed and validated, it can be used to virtually screen data base consisting of 3D multi-conformational structure of natural compounds 2. Additional virtual filtering tools for the profiling or absorption, distribution, metabolism and excretion parameters and toxicity might have an invaluable impact to aid a refined selection of virtual hits. Screening for related targets may help to increase the selectivity of the final virtual hits.
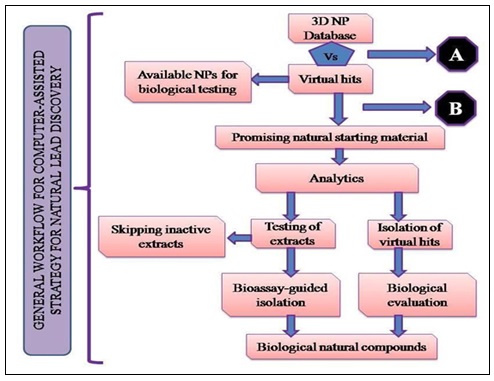
FIG. 1: GENERAL WORK FLOW FOR A COMPUTER ASSISTED STRATEGY FOR NATURAL LEAD DISCOVERY
- Futher In-silco Filters:
- ADME parameters
- Antitargets (toxicity)
- Off targets (selectivity)
- Additional beneficial targets
- Consensus hits
- Selection Criteria:
- Target related effect known for NP
- Target related effect known for natural material containing the predicted hit
- Empirical know-how (ethnopharmacology, folk medicine, bio-rationale)
How to Access Natural Products Bioactivity: The first isolation of a single chemical entity from natural pattern, that is morphine from opium, the latex from Papaver somniferum, by Friedrich Wilhelum Serturner in 1804 3 is one of the pharmacognosist’s goals to enhance the known chemical space by isolation of natural compound and establishing their structure. It provides interesting novel templates for synthetic chemists and serves as chemotaxonomic tool to support phylogenetic relationship. It provides a glimpse of structural requirement for an evolutionary trimmed bioactivity, allied target is usually unknown in first place. One of the most acknowledge procedure used to discover nature’s bioactivity is the exploration of curing or preventive agents traditionally employed by man, called Ethno- pharmacology 4 which is related to the study of ethnic groups and their use as drugs. It is distinctly linked to plant use. The biorational approaches can help staggeringly in the search for any bioactivity and still many more important findings of pharmaceutical interests. The differentiation between the traditional and modern drug discovery methods is made in Fig. 2A and 2B.
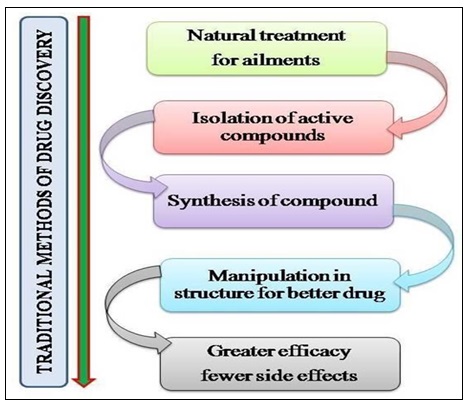
FIG. 2(A): DIFFERENCE BETWEEN THE TRADITIONAL METHODS AND MODERN METHODS OF DRUG DISCOVERY
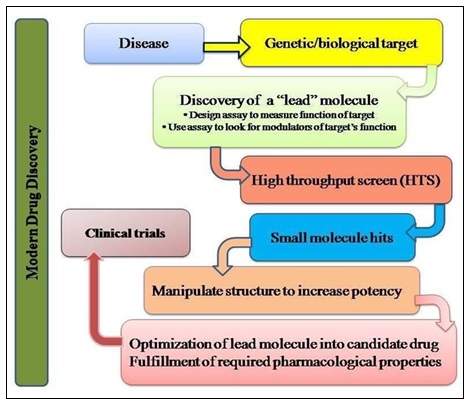
FIG. 2(B): DIFFERENCE BETWEEN THE TRADITIONAL METHODS AND MODERN METHODS OF DRUG DISCOVERY
Highly automated, miniaturized and very selective bioassays are used for screening compounds of natural and synthetic origin but also handle complex natural product preparation. The challenges however remain in the analysis and evaluation of the obtained results Fig. 3.
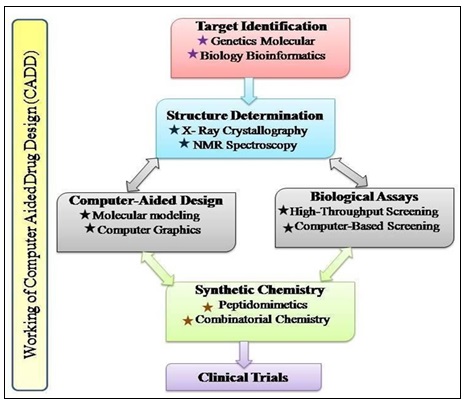
FIG. 3: WORKING OF CADD
The most challenging is the analyzing and evaluating these compounds, with the advent of computational techniques, there is an ever growing efforts to apply the anticipating in-silico power to the combined chemical and biological room in order to reorganize drug discovery, blueprint improvement and optimization. It has gained rapid popularity, implementation and appreciation 5.
TABLE 1: COMMONLY USED TERMS IN COMPUTATIONAL DRUG DESIGN
| Term | Explanation |
| Ligand | A substance (usually a small molecule) that forms a complex with a biomolecule to serve a biological purpose. In protein-ligand binding, the ligand is usually a signal-triggering molecule, binding to a site on a target protein. In DNA-ligand binding studies, the ligand is usually any small molecule or ion, or even a protein that binds to the DNA double helix. The binding occurs by inter molecular forces, such as ionicbonds, hydrogen bonds and vander Waals forces. Ligand binding to a receptor (receptor protein) alters its chemical conformation (three-
dimensional shape). |
| Binding affinity | The capability of ligand to form coordination bonds with receptor. The binding affinity of ligand with a receptor depends upon the force of attraction between the ligand and their receptor binding sites. Strong intermolecular force of attraction results to show high bonding affinity ligand binding, while the ligand binding of low- affinity involves lower and weak intermolecular force between ligand and their receptors. In other words, a ligand with high- affinity binding shows a long residence time at the receptor binding
site compared to low-affinity binding. |
| Inhibitory constant (Ki) | The concentration of the inhibit or that is required in order to Decrease the maximal rate of the reaction by half. The smaller the |
Lead Compound and Hurdles in Finding Lead Molecule: A lead compound is a small entity that works as the preliminary point for an optimization involving many small molecules that are closely related in structure. Many associations uphold databases of chemical compounds. Some of these are publically available but others are not. The database includes a vast number of compounds e.g. Database of American Chemical Society (ACS) contains approximately 10 million compounds. Spatial 3D (S3D) databases contain the information regarding the chemical and geometrical features including donor of hydrogen bond, acceptor of hydrogen bond, positive charge centers, aromatic ring centers, and hydrophobic centers.
The methods which provide an accurate prediction of which target a chemical entity may bind are in high demand, especially in research of natural products. There is a continuous growth in understanding of biological and physiological pathways and target of human-pathological relevance coming out of the human genome project. The increasing molecular knowledge of drug targets together with the advances made in biotechnology and bioassay are indispensable tools for discovery and development of more specific and selective drug. According to the modern western medicine, exploring a small compound for exactly one target that has to be blocked or stimulated and exclusively to abolish the focused impairment has been regarded as an ideal strategy for rational drug design 6.
Multi-target Compounds:
Problems and Challenges: Millions of compounds and drugs that are in clinical use or development have been found to be indiscriminate with regard to their biological targets and there effects can be anticipated originally 7. There is a distinct rise in late stages of attrition in phase 2 and 3; this can be mainly traced due to lack of efficacy and toxicology. These two criteria account for 30% failure. Because of increasing molecular insights with evaluation of post-genomic data explosion, we realize an imperfect knowledge of disease and complex biological system. There is an incomparable feasibility that an identified ligand also interacts with range of other macromolecules, which are so called multi-target compounds and can cause harmful side effects. A potent and highly selective drug affects a single gene in the cellular network, unwanted functions or pathways and alternative compensatory signaling routes can result in systems that are elastic against the arbitrary deletion and thus lead to futility. Although dealing with ligand of high binding affinity. Multi-target compounds open up additional medical use. Hitting more than one target might enable compound to be applied therapeutically in several unrelated diseases 8.
How to Access Multi-target Profile of Natural Products: In the research of natural products main core is now on determining the variety of targets that might be affected by a single chemical entity or even more complex to identify the variety of targets affected by each individual constituent of a multi-component natural preparation 9, 10. In order to venture the full therapeutic potential and reduce the toxicity, it’s imperative to identify a compounds respective target spectrum as through, as possible. The computational strategies might give valuable clues to search for bioactivities or to scrutinize toxic interactions and are very helpful for prioritizing experimental efforts.
Lipinski Rules for Intestinal Absorption (Rules of 5): Lipinski’s rule of five also known as the Pfizer’s rule of five or Rule of five (RO5), formulated by Christopher A. Lipinski in 1997, is employed to determine the drug likeness or determine whether a chemical compound having specific pharmacological activity, which will make it a likely orally active drug in humans. This rule is based on the observation that most orally administered drugs are small lipophilic substances. The rule describes molecular properties of drugs, which are vital for the pharmacokinetic profile for human use. If the compound is pharmacologically active, then the rule doesn’t predict it. As described in Lipinski's rule it is important to keep in mind that during the drug discovery when a pharmacologically active lead structure is optimized step-wise to increase its activity and selectivity and also ensure drug-like physicochemical properties are maintained. Candidate drugs that confirm to the RO5 tend to have lower attrition rates during clinical trials and hence have an increased chance of reaching the market 11.
Components of the Rule: Selection criteria for component as per the Lipinski rule includes: H-bond donors should be less than five; second rules states the sum of OH and NH group should be done, molecular weight should be less than 500 and Log P value should be less than 5.
Variants: In an attempt to improve the predictions of drug-likeness, the rules have spawned many extensions, for example: The partition coefficients log P should be in range -0.4 to +5.6, molar refractivity should be in-between 40-130, Molecular weight should be from 180 to 500, and number of atoms should be from 20 to 70. (Includes H-bond donors [e.g.: OH's and NH's] and hydrogen bond acceptors and the polar surface area should not be greater than 140 Ǻ.
Molecular Modeling: Helps us in predicting the biological activity of minute organic molecules by using calculations from computer. The in-silico modeling approaches uses recognized experimental data to calculate approximately the macromolecular binding capabilities, its geometry and similar chemical properties using various rule based and statistical computational techniques (knowledge based approaches). Specific aspect of binding can be understood with the help of quantum chemistry and laws of physics. The key aspect of all molecular modeling approaches is how the computer represents a chemical structure; that means which simplification are made for creating a computer representation and kind of models used 5.
Models for Chemical Structures: 2D, 3D and other Representations: Traditionally chemistry deals with 2D and 3D structures. A chemist is able to recognize important structural elements and functional groups in a way and develop new analogs from known active ligands using established bioisosteric replacement rules. The biochemical properties and the ability of ligand to interact with macromolecules depend on 3D form of molecule. The concept of3D molecule is much older than the use of computers in chemistry as it was already been introduced by Emil Fischer in 1890, he developed the famous lock and key metaphor, which compares the 3D complementarities between a macromolecular and a ligand to key fitting in to a lock. For computer-aided we have to decide whether to use topological (2D) information, use a 3D characterization of a molecule, or derive properties for either of these representations. The more data that are calculated for a molecular structure, the more computational power has to spend. Concerning the molecules, especially the 2D/3D conversion step, makes a huge difference in computational speed since conformations have to be generated. The ability of widely used programs to search conformational space has been extensively examined in recent years with good results 12, 13. Therefore, many 3D approaches resample conformations once and store the mina database for subsequent virtual screening 14. Fig. 3.
Knowledge Based Approaches: Knowledge -Based Approaches are the most frequently applied and most efficient methods in molecular modeling due to their universality. The aim of these methods is to find a representation of molecule that reflects all available knowledge that are gained through experiment, including structural data from x-ray or NMR experiments and structure-activity relationships from biological testing. Knowledge -based methods either use experimentally determined 3D macromolecular structure information (structure based design) or rely on the similarity of known active ligand (ligand-based design) 5.
Structure Based Design: Structure based design is depends on the accessibility of experimentally resolute structure of the macromolecule target under investigation. The Protein data bank (PBD) 15 represents the largest public repository of proteins and nucleic acid structures determined by x-ray crystallography or NMR. One of the reasons for increase in popularity of structure based approaches is the rapidly growing number of available coordinate files in the PBD and its illustrative nature 16.
The result of this shows the 3D arrangement of ligand bound molecule to a macromolecule and can be interpreted in an impulsive way 17. One of the important limits of crystal structure is that the atom coordinates are only models and the refraction pattern can be derived from x-ray crystallography only. Another important limitation is that all the macromolecules cannot be structurally determined 18.
Ligand Based Design: Ligand -based design is used in the cases when the structure of target is unknown or structure determination is impossible. The purpose of this model is to identify molecules that are similar to already known bioactive compounds. The problem of similarity can be approached from different perspectives, such as property similarity, descriptor similarity, or structural similarity. Many ligand based similarities exist, including topological fingerprints, shape based similarity, or methods that select several numeric molecular descriptors to fit statistical models.
Visualization: Rasmol, VMD, Molscipt, Raster 3D are tools used to optimize ligands or chemical compound and target molecule. Rasmol is used for depiction and exploration of biological macromolecule structure (computer program written for molecular graphics is used) 19.
Homology and Homology Modeling Programs: Homology modeling is used to predict the 3D structure of proteins. It is nothing but similarity searching for drug analogs. Molecular modeling is a science of interpretating the molecular structure with simulation of its attributes with the help of the equations of quantum and classical physics.
There are 2 computational tools for similar searching and sequence alignment such as BLAST, FASTA and for multiple sequence alignments Clastal W, Clastal X. There are 2 homology modeling programs. They are Swiss Model-makes it quick and easy to submit a target sequence and get back. Automatically, it generates a comparative model, which provides empirical structure with >30% sequence identity which exist to be used as a template 20.
Molecular Dynamics: It is a study of movement of molecule. Every molecule has its own frequency of vibration. Hence on increasing the potential energy, it can oscillate from position one to two through zero as energy at one and two positions is comparatively higher and least at zero position 21.
Energy Minimization: Also called energy optimization or geometry optimization; it is used to calculate the symmetric arrangement of molecules and solids. The final state of the system can be obtained by this technique which corresponds to minimum potential energy. In energy minimization one can acquire a molecule with zero energy state or least energy state. Equilibrium configuration is attained in this state. Energy minimization tools are GAMESS, Ghemical, PS13, and TINKER. Ghemical can be used or PS13 for quantum me Ghanical calculations. PyMol can be used to recognize the ligand binding pockets, together with the Deep View PDB viewer to investigate the amino acid sequences of the protein. To transfer files between programs, Open Babel may be constructive or even necessary to interconvert the file formats 22.
QSAR: QSAR can calculate the molecular property of a descriptor. It provides a wide variety of descriptors that you can use in determining new QSAR relationships. Limited number of datasets and little information regarding the validation was used by previous researchers, they recommended the use of SMILES or sdf files on a website to promote the calculation of additional parameters through other scientists. The self-organizing molecular field analysis (SoMFA) test set, represented the steroid set which can be used to build the initial comparative molecular field analysis (CoMFA), which can be downloaded from the website, Richards groups. This information aids a more-rapid evaluation of the SoMFA program 23.
Protein-Ligand Docking: Molecular docking predicts the binding mode of ligand to protein. The computational process is divided into two stages. In the 1st stage, a small organic molecule is placed into the binding site (pose placement). In the 2nd stage, all the available poses are ranked according to different algorithms (pose scoring). Various docking programs have been developed among them the most important ones are incremental ligand fragmentation and reconstruction (e.g., Flex X), volume- or shape- based algorithms (e.g., DOCK) and surface- based molecular similarity methods (e.g., Surflex 24. Scoring remains an unsolved problem, especially since the entropy part of the ligand binding energy is theoretically impossible to calculate from a single geometric snapshot of a ligand -protein complex.
Parallel Virtual Screening:
Activity Profiling: Molecular modeling technique can be used to predict the activity organic molecules to several targets or different isoforms in a single prediction step. The prerequisite for this approach is a collection of validated models for several targets. There are 3 main applications of activity profiling
- Identification of potential side effects by creating anti target models (e.g., for the hERG potassium channel or for some mechanistic cytochrome P450 inhibition).
- Optimization of isoform specificity for compounds (e.g., a specific kinase inhibition profile is crucial for the therapeutic applicability of a compound, so models for several kinases are built and screened in parallel).
- Target fishing (e.g., a newly synthesized compound or isolated natural product is screened against several models to determine which biological targets it should be tested against).
The most recent approaches for the multi-target profiling was published by, 7 who explored possible drug target effects computationally by using chemical similarities between 3,665 drugs and 65,241 ligands organized into 246 targets from MDL Drug Data Report (MDDR) database. The applied chemo-informatics methods provided tools to explore associations between drug-target effects and off-targets systematically, both to understand drug effects and to explore new opportunities for therapeutic intervention 7.
Software for Molecular Modeling:
- General purpose molecular modeling (large & small molecules) Molecular mechanics, dynamics and multifunctional programs.
- Quantum Chemistry calculations (small molecules) Molecular orbital or quantum mechanical calculations.
- Database of molecular structures (large & small molecules).
- Software for storage and retrieval of molecular structure data
- Molecular graphics (large & small molecules) Programs to visualize molecules
- QSAR (small molecules) and others
Challenges for the Application of Molecular Modeling to Natural Product Research:
Challenges from Chemo-informatic Point of View: Synthetic drugs show lesser molecular weight than the natural products. Natural drugs contain more oxygen atoms, fewer nitrogen atoms, three times more stereo centers, and their degree of unsaturation is higher than in synthetic drugs and they incorporate less aromatic rings. The major challenge for in-silico techniques is increased molecular weight and higher number of saturated bonds resulting in higher molecular flexibility. Too high degree of flexibility may also result in promiscuity, that is, a compound is fitted into a binding site in an implausible way, which stresses the need for visual inspection of virtual hits and selection of the most plausible virtual hits 25.
Availability of Natural Product Databases: Natural product molecular modeling is a compulsory tool for the databases of natural products. Such databases either contain compounds that are purchasable from commercial sources or known natural product from literature. Synthetic virtual hits that can be analyzed in terms of ethno-pharmacological and pharmacognostic context are less advantageous than virtual hits with known natural product sources and therefore they provide hints to related compounds or activities.
Commercial Natural Product Database: Several commercial suppliers of natural products and natural products derivatives offer downloadable structure-data files from their corporate websites. Analytic on provides the largest public collection of genuine natural products (www.analyticon-discovery.com). Smaller collection is available from BioFocus (www.biofocus.com), GreenPharma (www.greenpharma.com), SPECS (www.specs.com) and TimTech (www.timtech.net). VitasM (www.vitasmlab.com) provides approx. 25000 natural product derivatives.
Databases from Literature Sources: Chapman and Hall provided the most comprehensive databases of natural products i.e. the Dictionary of Natural Products (DNP) database which contains more than 214500 compounds and is updated every 6 months with the current literature data.
The DIOS database contains 9676 unique small molecular weight natural compounds based on information from ethno-pharmacological source.
The Natural Product Database (NDP) which was collected at the university of Innsbruck was last updated in 2009and currently contains 146,790 unique compounds from natural sources, including constituents of plants, animals fungi from land based as well as marine organism.
The Chinese Herbal Medicine Databases (CHMD) has 10,216, the Traditional Chinese Medicine Databases (TCMD) 10,458 compounds 10 and the Chinese Herbal Constituents Databases (CHCD), 27 7000 compounds from 240 Chinese herbs.
The Bioactive Plant Compounds Database (BPCD) also contains biological activity information for 78 targets, 2,597 compounds 28.
The Marine Products Database (MNPD) has 6000 chemicals from more than 10,000 marine derived materials.
Challenges from Pharmacognostic Point of View: There may be uncertainty surrounding the following issues
- Reliance on the previously published structure elucidation of the secondary metabolite.
- Correct entry in the data base, in many cases the configuration of a natural compound is not known or unambiguously solved.
- Is the aiming compound chemically and physically stable?
- Is it already known to be active on the scrutinized target? This can be seen in a positive sense as a proof of concept, or in negative sense, because its discovery is not a new finding anymore.
- Is it described or predicted to be toxic? What about its bioavailability?
- Is it commercially available (which applies to very rare cases, and usually they are very expensive)?
- Can the secondary metabolite rediscovered from there ported natural sources (how many reports are available about its isolation)?
- Might it be an art if actor a native natural material or only available intraces?
- Which is the best natural material or only accessible and legally available for collection?
- Are there conflicts with intellectual property rights or with the transfer of materials from outside biodiversity group?
Applications of Computers in Drug Design:
Anticancer agents: One of the major scientific endeavors of this century is the sequencing of the human genome. A major aspect of the utilization of this information will be the provision of tiny molecules which is able to acknowledge the selected sequence, perhaps with the goal of switching off particular genes as in cancer chemotherapy. Sometime antibiotics such as netropsin have been known to bind preferentially to sequences which are rich in A-T pairs. A variant supported this analysis has been to try to design a bioreductive ligand based upon netropsin. The notion of bioreductive anti-cancer agent stats with the very fact that tumors receive less blood and hence less oxygen than normal tissue. Therefore it becomes doable, at least principle, to contemplate having aligand which can exist in 2 forms i.e. oxidised and reduced, if the redox potential is suitable to be in the oxidized form in normal tissue however reduced in tumors. If solely reduced form binds to the macromolecular target, causes cell death, the discrimination in between the actions of cell causes destruction of normal cell, with concomitant reduced side effects. A second start line for sequence selective ligands is an organo-metallic molecule with chiral propertie. The propeller – like ruthenium tris-phenanthroline complexes also show differential binding between A-T and G-C sequences and what is more could exhibit a preference for purine 3', 5'pyrimidine sites in DNA 28.
Target Enzymes: If an enzyme structure is known then designing inhibitors which will block activity in the test- tube should be a relatively straight forward problem. It is more challenging if we add at the same time an attempt to make the ligand bioreductive as outlined above. The published work has taken dihydrofolate reductase as the target enzyme, but current activity is being centralized on thymidylate synthetase. Binding free energy of the inhibitor to the enzyme is a crucial quantity: strong binding is essential.
Drug Transport: Skeptics quite rightly point out that designing an enzyme inhibitor which will work in the test- tube is one thing; getting a compound which will work in a cell is another. Transport across the biological membrane is essential. Compounds must be soluble enough in the lipid to get into the membrane, but not so soluble that they remain there. Within the pharmaceutical industry the partition coefficient between water and n-octanol is used as a guide to membrane transport. The free energy perturbation technique just described can also be adapted to compute partition coefficients 29. More excitingly, however, it is becoming possible to model biological membranes. Starting with crystal structures of membranes involving DMPC (1,2-dimyristoyl-snglycero-3-phosphoryclholine) a highly realistic simulation is possible, involving a hydrated lipid bilayer. After very long molecular dynamics simulations the resulting membrane model is in agreement with all the available experimental data; lead up separation; order parameters and diffusion coefficients. This model can be used as the solvent in calculations of partition coefficients which should be considerably more realistic than experimental values in n-octanol. Furthermore it will be able to introduce cholesterol and protein into the model membrane to produce a true stimulation of how a given drug is transported into a cell 31.
Structure Determination of Proteins: One of the major contemporary scientific aims is to use the abundance of gene therefore protein sequences forecast the 3D structure of proteins going from primary to tertiary structure. The architecture of the binding site is known which would increase from handful of cases to numerous. Presently favored and only successful methods are all based upon finding resemblance and homologies among the protein of recognized sequence but unknown topology and known structures from 3D databases. Usually sequences are evaluated with scoring matrices being used to ascertain just how similar an undesired length of polypeptide in the unidentified is in assessment with a known case. One successful prediction of the important protein endothelin was made using the sequencing of amino acids. The property profile is smoother than an identity specification where each amino acid may be twenty. Where the similarity is low the use of coloured graphics is used to authorize the human eye to distinguish comparison which has many advantages although it is inevitably subjective. This advancement, by using CAMELEON, a computer program has recently been used to predict the structure of the interleukin-4 receptor. It’s predictable that the folding topology of the beta sheets of IL4R is the same as that seen in the crystal structure of 04, even though sequence individuality is less. Every domain of the IL4R
monomer was aligned with 0 4 using single residue hydropathy properties. Loops were added from a database of immunoglobulin’s so as to connect the sheets; side-chains rotamer library helped in adding side chains and unsolvated structure energy-minimized using molecular dynamics. The complete structure wasthus positioned in an 8A shell of water and unconstrained molecular dynamics carried out for 60 ps, lastly the complete structure was depreciated. Considering that the IL4 receptor acts in the same way as growth hormone receptor does, one molecule of IL4 was docked to a pair of receptor proteins. The docking shows which portion of IL4 joins to the receptor: especially D-helix. On the basis of this it should be possible to design kinetics of the crucial parts of the IL4 D-helix which would interfere with the biochemical consequence of the cytokine joining to its receptor, leading to antagonist with potential medicinal applications 31.
Biochemical Transformation: Computer aided techniques are feasible in those cases where no knowledge about the macromolecular target exist in atomic detail, and then it is still possible to this technique. An admired approach would be to compute the energy profile of a biochemical trans formation which would be popular to hinder, position the transition state or intermediate and then create a stable copy of these unstable transients familiar with the enzyme responsible for catalyzing the reaction and would hence operate as an inhibitor. Such a mimic has 2 rational steps that are necessary: find the transient structure and secondly blueprint a constant copy. The previous task is possibly best achieved by using a combination of quantum and molecular procedures. A current appraisal suggested the joined potential method used 32 for the trios phosphate isomerase reaction is possibly the procedure to be followed in the future. The second stage of the process appeals the beginning of the proposal of molecular similarity, a quantitative measure of just how similar one molecule is to another. Possibly the most imperative characteristic of resemblance is similarity of shape and secondly similarity of molecular electrostatic potential, equally can be presented by Gaussian functions which introduce major computational gabs in the calculation of similarity indices, of which several different types may be defined 29.
Molecular Similarity: Much more significant has been the achievement of similarity measures in structure reactivity relationships (SAR) and in quantitative structure-activity relationships (QSAR), 30 considered the series of steroids of which the binding affinity data was accessible and was a set studied in the earliest comparative molecular field 3-D structure activity toil.
The transverse confirmation correlation coefficients obtained from the statistical analysis balances well with those attained via frequently used matrices of similarities at grid points in the space surrounding the molecules which of come demand massive matrices of perhaps thousands of points. In addition there is no requirement for randomness about the coverage of molecular surface or the size of the three dimensional box into which the molecules are to be positioned Even though in its immaturity molecular similarity matrices seem to have a lot to offer in QSAR and in the optimization of molecular structures for particular biological effects.
Molecular Dissimilarity: Molecular dissimilarity can be defined as difference between a pair of molecule. Similarity has a range of values from zero to on. Identity is represented by unity. The importance of dissimilarity is the comparison of chiral forms of the same molecule. It can also be used as chirality coefficient, a number which gives a range of values of chirality rather than being an all or none property. Presently there is a great deal of research into producing pure chiral forms of compounds for the using it as a pharmaceutical agent, the supplementary active form being. Termed the eutomer and the less active the distomer, with their ratio being the eudysmic ratio. For a homologous series of compounds we have shown that there is a direct correlation between the eudysmic ratio and the chirality coefficient 31.
Concluding Remarks and Future Perspective: Natural compounds have huge impact in drug discovery and development, but still plenty of secondary metabolites are still needs to be discovered. Current research is benefited from a wealth of available experimental structural data and an endlessly growing number of therapeutically relevant macromolecular structures. In addition the increasing number of compounds isolated from natural resources, highlights the need to overcome the overwhelming amount of data and correlations. The use of computational techniques may prove effective in narrowing the search for the discovery of bioactive natural products and accordingly streamline the drug discovery process. In the last 2 decades, the application of predictive in-silico tools has emerged successful in identifying chemical entities having higher possibility of binding to the target molecules to elicit the desired biological response. In contrast to experimental screening, e.g. medium or high throughput screening, millions of compounds can be examined in-silico for their tendency to bind with the target proteins. The coordination of experts in the area of phytochemistry, pharmacognosy, pharmacology, and bioinformatics needs to be very crucial for a sensible application of data mining tool in this prospering field.
ACKNOWLEDGEMENT: Nil
CONFLICT OF INTEREST: Nil
REFERENCES:
- Drews J: Strategic trends in the industry. Drug Discovery Today 2003; 8: 411-420.
- Jose L and Medina-Franco L: Advances in computational approaches for drug discovery based on natural products. J Chem Inf Model 2013; 52: 95-104.
- Muller P, Paul N, Kellenberger E, Bret G and Rognan D: Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins 2004; 54: 671-680.
- Brown RD and Martin YC: The information content of 2D and 3D structural descriptorsrelevant to ligand-receptor binding. J Chem Inform Comput Sci 1997; 37: 1-9.
- Corrado T: Bioactive compounds from natural sources, natural products as lead compounds in drug discovery. Computational Approaches for the Discovery of Natural Lead Structures 2012; 4(2): 99-132.
- Chales RD and Martin YC: The information content of 2D and 3D structural descriptors relevant to ligand- receptor binding. J Chem Inform Comput Sci 1996; 37: 1-9.
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI and Hufeisen SJ: Predicting new molecular targets for known drugs. Nature 2009; 462: 175-U48.
- Hopkins AL, Besnard J, Setola GF, Abecassis K, Rodriguiz RM, Huang XP: Automated design fligand stopoly pharmacological profiles. Nature 2012; 492: 215-220.
- Kaminester LH, Pariser RJ, Pariser DM, Weiss JS and Shavin JS: A double-blind, placebo- controlled study of topical tetracaine in the treatment of Herpes labialis. J Am Acad Dermatol 1999; 41: 996-1001.
- Wang X, Cheng F, Li W, Wu Z, Zhang C and Li J: Prediction of polypharmacological profiles of drugs by the integration of chemical, side effect, and therapeutic space. J Chem Info Mod 2013; 753-62.
- Lipinski CA, Lombardo F, Dominy BW and Feeney PJ: Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 2001; 46: 3-26.
- Agrafiotis DK, Gibbs AC, Zhu F, Izrailev S and Martin E: Conformational sampling of bioactive molecules: A comparative study. JCIM 2007; 47: 1067-1086.
- Bostrom J, Norrby PB and Liljefors T: Conformational energy penalties of protein bound ligands. J Comput Aided Mol Dev 1998; 12: 383-396.
- Buckwalter JV, Chayavichitsilp P, Krakowski AC and Friedlander SF: Herpes simplex. Pediatr Rev 2009; 30: 119-129.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN and Weissig H: The protein data bank. Nucleic Acids Res 2000; 28: 235.
- Blundell TL: Structure based drugdesign. Nature 1996; 384: 23-26.
- Cottrel SJ, Gillet VJ and Taylor R: Incorporating partial matches within multi-objective pharmacophore identification. JCAMD 2006; 20: 735-749.
- Mestres J, Ekins S and Testa B: In-silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 2007; 152: 9-200
- Mofizur MD, Karim MdR, Ahsan MdQ, Khalipha ABR, Chowdhury MR and Saifuzzaman MD: Use of computer in drug design and drug discovery: a review. Int J Phar Life Sci 2012; 1(2): 1-21.
- Yoo J and Medina-Franco JL: Homology modeling, docking, and structure-based pharmacophore of inhibitors of DNA methyltransferase. JCAMD 2011; 25: 555-67.
- Timmers LF, Caceres RA, Dias R, Basso LA, Santos DS and Azevedo WF: Molecular modeling, dynamics and docking studies of Purine Nucleoside Phosphorylase from Streptococcus pyogenes. Biophy Chem 2009; 142: 7-16.
- Werner J and Geldenhuys: Optimizing the use of open-source software applications in drug discovery DDT 2006; 11: Number 3/4.
- Tetko IV: Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J Chem Inf Comput Sci 2002; 42: 1136-1145.
- Jain AN, Ruppert J and Welch W: Automatic identification and representation of protein binding sites for molecular docking. Prot Sci 1997; 6: 524-533.
- Wetzel S, Koch MA, Schuffenhauer A, Scheck M, Casaulta M and Odermatt A: Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proceedings of the National Academy of Sciences USA 2005; 102: 17272-17277.
- Ehrman TM, Barlow DJ and Hylands PJ: Phytochemical databases of Chinese herbal constituents and bioactive plant compounds with known target specificities. J Chem Inf Model 2007; 47: 254-263.
- Ehrman TM, Barlow DJ and Hylands PJ: Phytochemical informatics of traditional Chinese medicine and therapeutic relevance. J Chem Inf Model 2007; 47: 2316 -2334.
- Richards WG and Good AC: Structure- Activity Relationships from molecular similarity matrices. J Med Chem 1993; 36: 433.
- Reynolds CA, Burt C and Richards WG: A linear molecular similarity index. Quant Struct Act 1992; 11: 34.
- Goodford PJ: A computational procedure for determining energetically favorable binding sites on biological important macromolecules. J Med Chem 1985; 28: 849-857.
- Richards WG: Computer-aided drugdesign. Pure Appl Chem 1994; 66: 1589-1596.
- Baroni M, Costantino G, Cruciani D, Riganelli R and Clementi S: Generating optimal linear pls estimations (GOLPE): An advanced chemometric tool for handling 3D-QSAR problems. Quant Struct-Act Rel 1993; 12: 9-20.
- Sohpal VK, Dey A and Singh A: Computational approach for structure based drug design from a series of natural antiviral compounds for herpesviridae family. J Antivir Antiretrovir 2013; 5: 2-6
- Butler MS: The role of natural product chemistry in drug discovery. J Nat Prod 2004; 67: 2141-2153.
- Chen L, Morrow JK, Tran HT, Phatak SS, Du-Cuny L and Zhang SX: Structure-based drug design on protein targets. Curr Pharm Des 2012; 18: 1217-1239.
- Kuntz ID: Structure- based strategies for drug design and discovery Science 1992; 257: 1078.
- Veselovsky AV and Ivanov AS: Strategy of Computer-Aided Drug Design 2003; 33-40.
- Whittle PJ and Blundell TL: Protein Structure-Based Drug Design. Review Biophysics & Biomolecular Structure. 1994; 23: 349.
- Xiang ML, Cao Y, Fan WJ, Chen LJ & Mo YR: Computer aided drug design: Lead discovery and optimization. Comb Chem High Throughput Screen 2012; 15: 328-337.
- Ying Zu: Bioinformatics. Acta Pharmacologica Sinica 2008; 33: 4-16.
How to cite this article:
Chaurasia S: Unearthing of natural bioactives: an overview of computational approaches. Int J Pharmacognosy 2024; 11(8): 426-37. doi link: http://dx.doi.org/10.13040/IJPSR.0975-8232.IJP.11(8).426-37.
This Journal licensed under a Creative Commons Attribution-Non-commercial-Share Alike 3.0 Unported License.
Article Information
6
426-437
1495 KB
700
English
IJP
Simran Chaurasia
Department of Pharmaceutical Sciences and Technology, Maharaja Ranjit Singh Punjab Technical University, Badal Road, Bathinda, Punjab, India.
simranchaurasia777@gmail.com
06 August 2024
23 August 2024
28 August 2024
10.13040/IJPSR.0975-8232.IJP.11(8).426-37
31 August 2024