


NITRIC OXIDE AS A TARGET FOR HYPERTENSION MANAGEMENT: A REVIEW ARTICLE
HTML Full TextNITRIC OXIDE AS A TARGET FOR HYPERTENSION MANAGEMENT: A REVIEW ARTICLE
Alemayehu Toma * 1, Eyasu Makonnen 2 and Getnet Yimer 2
Department of Pharmacology 1, School of Medicine, Hawassa University, Hawassa, Ethiopia.
Department of Pharmacology 2, School of Medicine, Addis Ababa University, Addis Ababa, Ethiopia.
ABSTRACT: High blood pressure is well-established as a leading cause of stroke, ischemic heart disease, and cardiovascular disease. Globally cardiovascular disease accounts for approximately 17 million deaths a year, nearly one third of the total. A number of pharmaceutical agents are available for initial treatment of high BP. These include older molecules such as diuretics and beta-blocking agents, and newer molecules such as dihydropyridine calcium channel blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and direct acting vasodilators. Comprehensive hypertension management focuses on reducing overall cardiovascular risk and should be the preferred approach for initial management of hypertension. Nitric oxide is a gaseous signaling molecule that regulates various physiological and pathophysiological responses in the human body. Nitric oxide is a gaseous lipophilic free radical which reacts with various molecules to cause pleiotropic effects and it is generated by three distinct isoforms of nitric oxide synthase. The physiologic effects include circulation and blood pressure, platelet function, host defense, and neurotransmission in central nervous system and in peripheral nerves. The etiology of the association between impaired nitric oxide bioactivity and hypertension is complex, however, and has not been fully elucidated. Important mechanisms of hypertension and cardiovascular disease, in which impaired nitric oxide bioactivity plays a major role, include arterial stiffness and increased systolic pulse wave velocity, and possibly chronic sympathetic nervous system activation to be investigated in-depth. This molecule plays a vital role in a wide variety of pathophysiological and biochemical reactions. Nitric oxide could be a critically important mediator in pathophysiology of different cardiac diseases. Nitric oxide has been identified as one of the key targets for novel therapeutic interventions to minimize irreversible tissue damage associated with complications of hypertension and other cardiovascular disorders.
| Keywords: |
Hypertension, nitric oxide, Signaling molecule, and cardiovascular disorders
INTRODUCTION: High blood pressure is well-established as a leading cause of stroke, ischemic heart disease, and cardiovascular disease.
Globally cardiovascular disease accounts for approximately 17 million deaths in the year 2013, nearly one-third of the total. Of these, complications of hypertension account for 9.4 million deaths worldwide in 2013. Hypertension is responsible for at least 45% of deaths due to heart disease, and 51% of deaths due to stroke 1.
There is, therefore, a need to lower BP in all groups of patients. This can be achieved by non-pharmacological (lifestyle measures) as well as pharmacological means. Lifestyle changes include dietary interventions, weight control, tobacco cessation, exercise, and stress management. Some pharmaceutical agents are available for initial treatment of high BP. These include older molecules such as diuretics and beta-blocking agents, and newer molecules such as dihydropyridine calcium channel blockers (CCB), angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), and direct acting vasodilators. Comprehensive hypertension management focuses on reducing overall cardiovascular risk and should be the preferred approach for the initial management of hypertension 2.
The role of nitric oxide (NO) in the cardiovascular system and regulation of blood pressure has been studied extensively. NO is continuously synthesized from the amino acid L-arginine by endothelial cells, utilizing the constitutive Ca 2+/calmodulin-dependent enzyme NOS. The principal physiological stimulus for NO synthesis and release from the endothelium is probably the shear stress of blood flowing over the surface of the vessel by a non-receptor-dependent mechanism. NO, released from the endothelium as a gas or attached to other molecules, stimulates soluble guanylate cyclase in vascular smooth muscle underlying the endothelium, producing increased concentrations of cGMP, thus activating cGMP-dependent kinases that promote relaxation 3.
Endothelial dysfunction, which is defined by decreased endothelium-dependent vasodilation, is associated with an increased number of cardiovascular events. NO bioavailability is reduced by altered endothelial signal transduction or increased formation of O2− (oxygen superoxide radical) reacting with NO to produce ONOO (peroxynitrite). Endothelial dysfunction is therapeutically reversible and physical exercise, calcium channel blockers, ACEIs (angiotensin-converting enzyme inhibitors) and angiotensin receptor antagonists improve flow-evoked endothelium-dependent vasodilation in patients with hypertension 4.
Nitric oxide (NO) is a gaseous signaling molecule that regulates various physiological and pathophysiological responses in the human body. NO is a gaseous lipophilic free radical which reacts with various molecules to cause pleiotropic effects, and it is generated by three distinct isoforms of nitric oxide synthases (NOS). The physiologic effects include circulation and blood pressure, platelet function, host defense, and neuro-transmission in the central nervous system and peripheral nerves 3, 4.
NO acts as a neurotransmitter in non-adrenergic noncholinergic (NANC) nerves and regulates smooth muscle tone. NO is also involved in the host defense in bronchial epithelium, and it acts as an inflammatory mediator in pathological states. Inhaled NO may have clinical implications in certain conditions as a bronchodilator and vasodilator, and NOS inhibitors are believed to be of benefit in inflammatory lung diseases. Also, exhaled NO can be measured as a marker of asthma and other inflammatory lung diseases. In this review, we discuss the role of NO in hypertension as well as the targets of NO 5.
Biosynthesis of Nitric Oxide: NO is synthesized from L-arginine in a reaction catalyzed by a family of nitric oxide synthase (NOS) enzymes. The three NOS isoforms have similar enzymatic mechanisms that involve electron transfer for oxidation of the terminal guanidine nitrogen of L-arginine Fig. 1. These enzymes all require several cofactors for proper function, including tetrahydrobiopterin (BH4), nicotinamide-adenine dinucleotide phosphate (NAD-PH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and iron protoporphyrin IX (haem). Most of the effects of NO, on smooth muscle cells, platelets, and cardiac myocytes, are mediated through its activation of soluble guanylate cyclase (cGMC) and amplifying the production of cyclic guanosine monophosphate (cGMP), but the increasing evidence indicates that NO mediates its effects also, through cGMC-independent mechanisms. Active NOS is a tetramer formed by two NOS proteins and two calmodulin molecules 4, 6.
Three different NOS isoforms have been characterized. The neuronal NOS (nNOS, NOS I) is predominantly expressed in neurons in the brain and peripheral nervous system. Endothelial NOS (eNOS, NOS III) is mainly expressed in endothelial cells. Both nNOS and eNOS are constitutively expressed and are inactive in resting cells. Increase in free intracellular calcium concentration ([Ca2+]i) stabilizes the binding of calmodulin to eNOS and nNOS, and activates the enzyme to produce NO. Stimuli that increase the [Ca2+]i (e.g. acetylcholine in endothelial cells) trigger the production of NO, and when the [Ca2+]i decreases, the NO production ceases. The regulations make NO production by constitutively expressed NOSs transient and short lasting 5, 6.
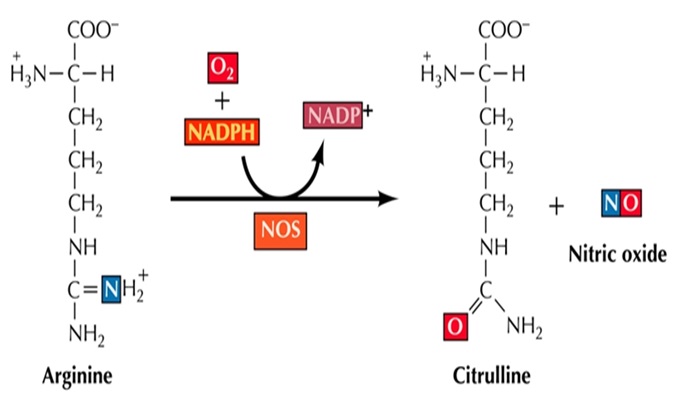
FIG. 1: BIOSYNTHESIS OF NO FROM L-ARGININE
The third isoform of the NOS family is the inducible NOS (iNOS, NOS-II). No iNOS expression is found in most resting cells. Exposure to microbial products, such as lipopolysaccharide (LPS) and dsRNA or proinflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) induces the expression of iNOS gene in various inflammatory and tissue cells. Binding of calmodulin to iNOS is tight even at low [Ca2+]i, and therefore, iNOS is also called as a calcium-independent NOS, and it can constantly produce high levels of NO for prolonged periods 5, 6.
Biochemical Effects of Nitric Oxide: NO reacts exclusively with other paramagnetic species, such as other radicals or metal centers, due to the presence of an unpaired electron. It can react with reactive oxygen species (ROS) such as superoxide anion (O2-) to form more active intermediate, such as peroxynitrite, in reaction that is six times faster than the dismutation of O2 by superoxide dismutase (SOD). Peroxynitrite directly oxidises cysteine and tryptophan, while modification of lysine and arginine probably occurs via secondary reactions with lipid hydroperoxide radicals. NO, unlike other intracellular molecules, is freely diffusible and influences a number of biosynthetic, metabolic, signaling and membrane transport processes. NO dilates the vascular tree and inhibits platelet aggregation, thrombus formation, leukocyte adhesion and vascular proliferation. Exogenous and endogenous NO inhibits vascular smooth muscle cell (VSMC) proliferation and migration. Reduced NO bioavailability is implicated in the development of vascular diseases, although it is poorly understood whether this is a cause of, or result of endothelial dysfunction (ED) or both pathogenic events 6, 7.
The existing knowledge on the role of NO in physiological and pathophysiological states has opened up a wide range of possibilities, especially in understanding of the mechanisms of actions of drugs that modulate NO action. The high level of inducible NO is thought to be a major factor in the severe hypotension that characterizes the toxic shock syndrome. The role of NO and NOS in regulating vascular physiology, through neuro-hormonal, renal and other non-vascular pathways, as well as direct effects on arterial smooth muscle, appear to be more intricate than was originally thought 7.
Impaired Nitric Oxide and Hypertension: Endothelial dysfunction is often described in individuals with hypertension. Multiple studies of flow-mediated dilatation of the brachial artery have shown that patients with hypertension exhibit blunted arterial vasodilation in response to endothelium-dependent vasodilators, such as Ach, while vasodilatory responses to endothelium-independent vasodilators, such as sodium nitroprusside, are preserved 8.
Role of Nitric Oxide in Endothelial Dysfunction: The importance of NO in promoting endothelial homeostasis is demonstrated by the association between impaired NO bioactivity and endothelial dysfunction, which is characterized by the imbalance of endothelium-derived vasoconstrictive and vasodilatory substances, with a shift toward greater vasoconstriction, inflammation, and thrombosis 7, 8.
One proposed mechanism of impaired NO availability and endothelial dysfunction is oxidative stress. Free radical molecules function normally as signals to modulate vascular tone. Oxidative stress exists when pro-oxidant processes exceed the capacity of antioxidant mechanisms to maintain an appropriate balance. Oxidative stress is produced with increased production of ROS, including superoxide anion that is derived from xanthine oxidase, cyclooxygenase, and NADPH oxidase enzyme systems. ROS react with NO forming peroxynitrite, thereby decreasing NO bioactivity. Oxidative stress may cause a deficiency of L-arginine and tetrahydrobiopterin (BH4). BH4 is an essential cofactor of NOS, along with NADPH, Ca2+ ⁄calmodulin, and flavin nucleotides. Reduced availability of BH4 causes eNOS to produce superoxide instead of NO, a process known as NOS uncoupling; this results in increased formation of peroxynitrite, a highly reactive oxidant that can be highly toxic. In certain circumstances, however, peroxynitrite has been reported to have beneficial effects in the cardiovascular system; therefore, the biologic effects of peroxynitrite can be paradoxical 8, 10.
However, these apparently contradictory responses could well be due to the environment in which studies were conducted. The maleficence action may be due to the conversion of peroxynitrite into deleterious mediators with hydroxyl radical-like activities, whereas, the beneficial action of peroxynitrite may be due to reaction of peroxynitrite with plasma, red cell glutathione, or plasma cysteine and albumin, with the production of NO or an NO donor–like compound. Peroxynitrite induces accumulation of cGMP in a glutathione-dependent manner in endothelial and smooth muscle cells, and peroxynitrite produces thiol-dependent stimulation of purified guanylate cyclase 8, 9.
Most commonly, ED is characterized by an impaired NO bioavailability due to reduced production of NO by NOS or increased breakdown by ROS and it occurs early in the development of atherosclerosis. Cardiovascular risk factors are often associated with ED, which is also prognostic for the occurrence of cardiovascular events. ED links cardiovascular disease because the ED plays a major role in the development of cardiovascular disease. Endothelium-dependent vasodilatation is mediated by NO, prostacyclin, and an endothelium-derived hyperpolarising factor (EDHF), and involves small and intermediate conductance Ca (2+)-activated K(+) channels. The opening of small and intermediate conductance Ca (2+)-activated K(+) channels is associated with endothelium-derived hyperpolarising factor-type vasodilatation, but, through increased endothelial cell Ca(2+) influx, L-arginine uptake, and decreased ROS production, it may also lead to increased NO bioavailability and endothelium-dependent vasodilatation. Therefore, small and intermediate conductance Ca (2+)-activated K (+) channels may be drug targets for the treatment of endothelial dysfunction in cardiovascular disease 9, 11.
The eNOS mostly produce NO produced by three different isoforms of NOS widely expressed in virtually all vascular cell types in endothelial cells where it plays a crucial role in vascular tone and structure regulation. It also exerts an anti-inflammatory influence, inhibits platelets adhesion and aggregation, and prevents smooth muscle cells proliferation and migration. Because of this, the loss of NO production and bioactivity could explain why diverse pathological conditions such as hypercholesterolemia, hypertension, diabetes, and cigarette smoking are all considered risk factors for atherosclerosis 3, 10.
NO is released by endothelial cells mainly in response to shear stress, but also by many other molecules such as acetylcholine, bradykinin, thrombin, adenosine diphosphate (ADP), phosphodiesterase type 5 (PDE5) inhibitors and nitro vasodilators among others, leading to a relaxation of VSMC. There are also exogenous sources of NO that could influence its availability via nitrites, S-nitroso thiols, N-nitroso proteins, and iron-nitrosyl complexes. The endothelium possesses all three NOS isoforms, eNOS, nNOS and under certain conditions, for example, inflammation, can express iNOS. eNOS is only fully functional in a dimeric form, and the functional activity of the eNOS dimer is dependent on the number of bound tetrahydrobiopterin (BH4) molecules. the eNOSs system mainly contributing to the endothelium-derived hyperpolarizing factor /H2O2 responses in microvessels while serving as a NO-generating system in large arteries 5, 11.
Recent Advances in Pharmacology of Nitric Oxide in Hypertension: There are many inhibitors of biological activity of NO, such as monocytes, decreased L-arginine uptake, decreased co-factors (Ca2+, calmodulin, BH4), inhibition of electron flow (nicotinamide adenine dinucleotide phosphate-oxidase (NADPH), flavins), inhibition of NOS expression, inhibition of substrate binding to NOS and NO scavengers. Also increased ROS concentrations (e.g. superoxide anion) reduce the amount of bioactive NO and form toxic peroxynitrite. Peroxynitrite in turn, can "uncouple" endothelial NO synthase to become a dysfunctional superoxide-generating enzyme that contributes to vascular oxidative stress. Beside them, some oxidation products of NO with ROS and thiols, such as nitrite, S-nitroso thiols and N-nitroso proteins are nowadays regarded as physiological storage pool of NO since these reactions are reversible and this can change an image of NO being paracrine factor. Among all of them, a lot of attention in recent years is focused on BH4 bioavailability and its role as an essential cofactor for optimal NOS production of NO 7, 12.
BH4 is a critical cofactor for all three isoforms of NOS and is involved in the reduction of the heme iron of the enzyme to ultimately form an iron-oxy species that hydroxylates L-arginine to produce NO. In the absence of BH4, the NOS enzymes produce superoxide rather than NO, a situation referred to as NOS uncoupling. Oxidation of BH4 leads to eNOS uncoupling in a variety of other disease states including atherosclerosis, myocardial infarction, and diabetes. Recent in vitro studies with purified eNOS, implicated it as an important source of vascular ROS production. However, in the intact cell, BH4 depletion alone does not appear to be sufficient of an insult to trigger eNOS uncoupling. Increased levels of the BH4 oxidation product dihydrobiopterin (BH2), rather than BH4 depletion alone, is the molecular trigger for NO insufficiency and eNOS uncoupling. As such, there are three states of eNOS in regard to the biopterin cofactor: BH4-eNOS (the functional NO synthase), BH2-eNOS, and biopterin-free eNOS, both of which are uncoupled- eNOS, which have oxidase activity 9, 10.
It is becoming increasingly clear that oxidative stress and perturbation of redox equilibrium in the endothelium, are of central importance for NOS activity and NO production and that it can be seen and evaluated by redox balance of biopterin cofactors. One mechanism of BH4 oxidation is through laminar shear stress that acutely stimulates endothelial production of NO and over the long term enhances eNOS gene expression 10, 13.
It has been shown that BH4 deficiency and NOS uncoupling likely contribute to the vascular inflammation and abnormal cytokine milieu induced by disturbed flow without affecting systemic immune cell numbers. The other mechanism of BH4 oxidation and subsequent NOS uncoupling is via O2_ produced by NADPH oxidases activated through oscillatory shear stress. ROS produced by uncoupled NOS could further activate NADPH oxidases, in a feed-forward fashion and that could contribute to ROS production throughout the vessel wall. Oral BH4 supplementation prevented NOS uncoupling and improved endothelial function in the carotid exposed to disturbed flow induced by partial carotid ligation. These findings highlight a pivotal role of BH4 deficiency and NOS uncoupling in atherosclerosis progression, particularly under the patterns of low and oscillatory shear flow, and indicate that modulation of vascular BH4 levels could be a therapeutic target for preventing atherosclerosis at branches and curvatures in the arterial tree 10, 14.
Studies in humans and laboratory animals have shown that the shear stress induced by physical exercise is a powerful stimulant for the release of vasorelaxing factors produced by the vascular endothelium, such as NO and the EDHF, resulting in a decrease of arterial pressure levels. There is extensive evidence that thiols potentiate eNOS activity and alleviate oxidative stress. NOS uncoupling induces oxidative stress and has previously been shown to occur with depletion of L-Arginine or BH4 and elevation of methylarginine levels 13.
It has been reported that eNOS possesses specific redox-sensitive thiols that are readily S-glutathionylated in endothelial cells and vessels with marked endothelial dysfunction and hypertension. This oxidative modification switches eNOS from its classical NO synthase function to that of an NADPH-dependent oxidase generating oxygen free radical. Because NO and O2- have many opposing roles in cell signalling and vascular function, S-glutathionylation of eNOS will trigger profound changes in cellular and vascular function and will mediate redox-signalling under oxidative stress. Therapeutics with thiol-reducing properties can therefore now be developed and refined as potent drugs for reversing ED and ameliorating hypertension and other cardiovascular disease. Therapeutically, drugs in clinical use such as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II type 1 (AT1) receptor blockers, and statins have pleiotropic actions that can improve endothelial function 14, 15.
Congestive heart failure (CHF) is a complex clinical syndrome that can result from any structural or functional cardiac or non cardiac disorder that impairs the ability of the heart to respond to physiological demands for increased cardiac output. The role of NO in CHF is complex. On the one hand, lack of NO is leading to ED with its detrimental consequences including impaired tissue perfusion, myocardial ischaemia, and vascular remodeling. On the other hand, higher concentrations of NO, which have been observed in the failing myocardium, may cause the loss of myocytes and inhibit myocyte contractility. High quantities of NO released during septicemia result in cardiovascular collapse and eventual death 16.
Other studies have also reported increased myocardial iNOS expression and activity in CHF. Indeed, because high concentrations of NO attenuate myocyte contraction and catecholamine responses, the one proposed mechanism of myocardial dysfunction in CHF is excessive NO production secondary to increased inflammatory cytokines. In support of this concept, studies have shown that NOS blockade improves myocardial beta-adrenergic responsiveness.
Recent investigations have shown that in failing myocardium, chronic beta-adrenergic blockade improves myocardial function and left ventricular remodeling, although the cellular mechanisms responsible for these salutary effects have not been fully defined. Also, for example, in dogs with pacing-induced cardiac failure, a reduction in O2 consumption has been observed, and consequently, in coronary flow, suggesting a down-regulation of energy metabolism. It has also been seen that in dogs with heart failure (HF), selective iNOS inhibition with S-methyl-isothiourea increased left ventricular contractility and oxygen consumption at rest and during exercise, indicating that, unlike what is observed in normal hearts, in failing hearts NO in excess is mainly produced by iNOS rather than by eNOS 17, 18.
The increase in coronary blood flow after inhibition of the release of NO is clearly due to the prevalence of the effect mediated by the increased oxygen consumption on the vessels that would be otherwise counteracted by a reduced concentration of NO in the vascular wall. It was shown that the reduction in O2 consumption, which may take place in CHF, depends on the increased availability of NO. It has been shown that NO has a negative effect on the beta-adrenergic response of ventricular myocardium that appears to be enhanced in failing myocardium. Moreover, beta-adrenergic stimulation by isoproterenol increases NO release and can amplify its depressant modulation 19, 20.
It may then be argued that in HF the effect of an increased beta-adrenergic activation on myocardium is attenuated by a sort of negative functional feedback. Since, a long-lasting beta-adrenergic activation is a maladaptive phenomenon, this feedback can play a protective role against the progression of CHF. Carvedilol, a nonselective beta blocker with antioxidant and alpha adrenergic receptor blocking activities, is one of the most effective beta blockers in reducing ventricular tachyarrhythmias and mortality in individuals with HF. The reports on the effect of carvedilol on NO production are scarce yet controversial. It was reported that carvedilol stimulated the expression of iNOS in cardiac myocytes. Also, one in vivo study using rats indicated that the drug decreased arterial pressure while it increased NO production 21, 22.
In the clinical setting, the effects of carvedilol on NO may be diverse depending on the local concentrations of free radicals and NO. The proposed antioxidant mechanisms of carvedilol include: [1] direct interaction with oxygen radicals; [2] prevention of the depletion of intracellular antioxidants; [3] attenuation of iron-mediated free radical formation 23.
The antioxidant and alpha-blocking activities of carvedilol have been suggested to contribute to its beneficial effects, but clinical studies have not supported the benefits of antioxidants and alpha blockers. It was indicated that carvedilol is the only beta blocker tested that can effectively suppress arrhythmogenic store overload-induced Ca2+ release that can lead to triggered arrhythmias and sudden death. Carvedilol could act by directly altering the function of channel ryanodine receptors 2, independent of its beta- or alpha-blocking or antioxidant activities. Regarding other antioxidants and their potential role in ameliorating increased content of NO and ROS in CHF, no trials have been identified on morbidity in patients with CHF. One trial of 56 patients with advanced heart failure indicated that supplementation with vitamin E did not result in any significant improvements in prognostic or functional indexes of heart failure or quality of life 24, 25.
Future Directions: Hypertension is increasingly understood to be a complex disorder that is strongly associated with other risk factors for cardiovascular disease. Clinical studies have demonstrated that patients with hypertension have a reduced vasodilatory response to endothelium-dependent vasodilators, such as acetylcholine, and that blockade of NOS also blunts endothelium-dependent vasodilation 26. Also, there is evidence for a role of eNOS polymorphisms, and experimental studies suggest a possible genetic component to impaired NO bioavailability and hypertension remains further investigation.
The etiology of the association between impaired NO bioactivity and hypertension is complex, however, and has not been fully elucidated. Important mechanisms of hypertension and cardiovascular disease, in which impaired NO bioactivity plays a major role, including arterial stiffness and increased systolic pulse wave velocity, and possibly chronic sympathetic nervous system activation to be investigated in-depth.
The overexpression of iNOS and the increased production of NO seem to characterize the congestive heart failure and other cardiovascular disorders. This is accompanied by an increased ROS production, which may lead to ONOO (peroxynitrite) formation. NO has a negative impact on the beta-adrenergic response of ventricular myocardium that is enhanced in failing myocardium 27, 28. But the exact mechanism behind the effect needs further exploitation.
Further clarification of the role of impaired NO bioactivity in the pathogenic mechanisms of hypertension and cardiovascular disorders could have important implications for the management of hypertension and cardiovascular diseases.
CONCLUSION: Increasing knowledge of the role of iNOS in the heart has stimulated efforts to target NO pathway pharmacologically. NO is a very important messenger molecule so far as its spectrum of actions is concerned. This molecule plays a vital role in a wide variety of pathophysiological and biochemical reactions. NO could be a critically important mediator in the pathophysiology of different cardiac diseases. NO has been identified as one of the key targets for novel therapeutic interventions to minimize irreversible tissue damage associated with complications of hypertension and other cardiovascular disorders.
ACKNOWLEDGEMENT: Nil
CONFLICT OF INTEREST: Nil
REFERENCES:
- World Health Organization. World Health Report 2013: Reducing risks, promoting a healthy life. http://www.who.int/whr/2013/2014. Accessed January 2014.
- Branislava D, Katarina S, Sanja S, Hans-Dirk D and Esma R: Nitric oxide and its role in cardiovascular diseases. The Open Nitric Oxide Journal 2011; 3: 65-71.
- Simonsen U, Rodriguez-Rodriguez R, Dalsgaard T, Buus, N and Stankevicius E: Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol 2009; 61: 105-115.
- Brunner H, Cockcroft JR and Deanfield J: Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. J Hypertens 2005; 23: 233-246.
- Uppu RM, Nossaman BD, Greco AJ, et al. Cardiovascular effects of peroxynitrite.Clin Exp Pharmacol Physiol 2007; 34: 933-937.
- Zalba G, Beaumont FJ and San-Jose G: Vascular NADH⁄ NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2008; 35: 1055-1061.
- Schmidt HH, Pollock JS and Nakane M: Ca2+⁄ calmodulin-regulated nitric oxide synthases. Cell Calcium 1992; 13: 427-434.
- Nossaman BD and Kadowitz PJ: Potential benefits of peroxynitrite. Open Pharmacol J 2008; 2: 31-53.
- Podjarny E, Hasdan G and Bernheim J: Effect of chronic tetrahydrobiopterin supplementation on blood pressure and proteinuria in nephrectomized rats. Nephrol Dial Transplant 2004; 19: 2223-2227.
- Fortepiani L and Reckelhoff J: Treatment with tetrahydrobiopterin reduces blood pressure in male SHR by reducing testosterone synthesis. Am J Physiol Regul Integr Comp Physiol 2005; 288: R733-R736.
- Thomas D, Gary E, Bobby D, Nossaman P and Kadowitz J: Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. Journal of clinical hypertension 2012; 14: 198-207.
- Christopher G and David J: Review focus on inorganic nitrite and nitrate in cardiovascular health and disease. Cardiovascular Research 2011; 89: 489-491.
- Philip N, Paulo I, Ciara O, Giuseppe D, Colin M and Mauricio S: Exercise and possible molecular mechanisms of protection from vascular disease and diabetes: the central role of ROS and nitric oxide. Clinical Science 2010; 118: 341-349.
- Stephan H, Christian M, Ulrich L and Michael B: Nitric oxide-donating statins: a new concept to boost the lipid-independent effects. Cardiovascular Research 2012; 94: 395-397.
- Ulrich F and Huige L: Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. British Journal of Pharmacology 2011; 164: 213-223.
- Paulino R, Emanuele G and Eliseu S: Nitric oxide synthase expression correlates with death in an experimental mouse model of dengue with CNS involvement. Virology Journal 2013; 10: 267-276.
- Taylor C, Sarah D and Kelly M: L-Arginine supplementation in type ii diabetic rats preserves renal function and improves insulin sensitivity by altering the nitric oxide pathway. International Journal of Endocrinology; Article ID 171546,7 pages http://dx.doi.org/10.1155/2014/171546 Accessed in January 2014
- Mahmud A and Feely J: Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension 2005; 46: 1118-1122.
- Davey-Smith G, Lawlor DA and Harbord R: Association of C-reactive protein with blood pressure and hypertension: life course confounding and mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol 2005; 25: 1051-1056.
- Betz B, oller-Ehrlich K and Kress T: Increased symmetrical dimethylarginine in ischemic acute kidney injury as a causative factor of renal L-arginine deficiency. Translational Research 2013; 162: 67-76.
- Aiello S, Remuzzi G, and. Noris M: Nitric oxide/endothelin balance after nephron reduction. Kidney International Supplements 1998; 65: S63-S6.
- Toba H, Sawai N and Morishita M: Chronic treatment with recombinant human erythropoietin exerts renoprotective effects beyond hematopoiesis in the streptozotocin-induced diabetic rat. European Journal of Pharmacology 2009; 612: 106-114.
- Tunctan B. Korkmaz B and Sari A: Contribution of iNOS/sGC/PKG pathway, COX-2, CYP4A1, and gp91phox to the protective effect of 5, 14-HEDGE, a 20-HETE mimetic, against vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock. Nitric Oxide 2013; 33: 18-41.
- Morris M, Gao J, Cooper T, Kepka-Lenhart D and Awad A: Arginase-2 mediates diabetic renal injury. Diabetes 2011; 60: 3015-3022.
- Bellinghieri G, Santoro D, Mallamace A and Savica V: L-arginine: a new opportunity in the management of clinical derangements in dialysis patients. Journal of Renal Nutrition 2006; 16: 245-247.
- Di-Paola R, Impellizzeri D and Mondello P: Palmitoylethanolamide reduces early renal dysfunction and injury caused by experimental ischemia and reperfusion in mice. Shock 2012; 38: 356-366.
- Peeri M, Habibian M, Azarbayjani A and Hedayati M: Protective effect of aerobic exercise against L-name-induced kidney damage in rats. Archives of Industrial Hygiene and Toxicology 2013; 64: 43-49.
- Awumey E, Bridges L, Williams L and Diz D: Nitric-oxide synthase knockout modulates Ca2+-sensing receptor expression and signaling in mouse mesenteric arteries. The Journal of Pharmacology and Experimental Therapeutics 2013; 346: 38-47.
How to cite this article:
Toma A, Makonnen E and Yimer G: Nitric oxide as a target for hypertension management: a review article. Int J Pharmacognosy 2014; 1(5): 288-95. doi: 10.13040/IJPSR.0975-8232.1(5).288-95.
This Journal licensed under a Creative Commons Attribution-Non-commercial-Share Alike 3.0 Unported License.
Article Information
2
288-295
506
2106
English
IJP
A. Toma *, E. Makonnen and G. Yimer
Department of Pharmacology, School of Medicine, Hawassa University, Hawassa, Ethiopia.
alexpharma99@yahoo.com
14 March 2014
16 April 2014
28 April 2014
http://dx.doi.org/10.13040/IJPSR.0975-8232.1(5).288-95
01 May 2014