


ISOLATION AND CHARACTERIZATION OF ACTIVE CONSTITUENTS FROM PLANT PISONIA ACULEATA LINN BY SPECTRAL ANALYSIS
HTML Full TextISOLATION AND CHARACTERIZATION OF ACTIVE CONSTITUENTS FROM PLANT PISONIA ACULEATA LINN BY SPECTRAL ANALYSIS
Shweta P. Ghode *, Prashant D. Ghode, Vibhavari M. Chatur and Rohini Kolhe
Department of Pharmacognosy, Rasiklal M. Dhariwal Institute of Pharmaceutical Education and Research, Chinchwad, Pune - 411019, Maharashtra, India.
ABSTRACT: Botanicals and herbal preparations for medicinal usage contain various types of bioactive compounds. The focus of this paper is on the analysis of bioactive compounds present in the Pisonea aculeata Linn. having anticancer activity involving the applications of chromatographic techniques such as TLC and column chromatography, Spectrophotometric techniques such as UV, 1H-NMR, 13C-NMR, and EIMS spectral studies. The present investigation was carried out by fractionating the Methanolic extract of Pisonia aculeata (MPA) using benzene, diethyl ether, and ethyl acetate. Ethyl acetate fraction on concentration subjected to separation and purification on column chromatography. Fractions were collected & homogeneity was examined on TLC. A Fraction 77-90 on concentration yielded a pure yellowish homogeneous solid and gave dark green coloration with neutral ferric chloride and violet coloration with Molish’s reagent. In order to find out the nature of the glycoside, compound I was subjected to acid hydrolysis. The basic flavonoid structure of the compound studied by recording UV spectrophotometer showed an intense UV maxima at 273 nm (band II) and 329 nm (band I), indicating the flavone nature of it. The 1H-NMR spectrum of compound I was recorded using AMX 400 (400 MHz) spectrometer using DMSO-d6 as the solvent showed a pair of doublets in the aromatic region at d 7.89 ppm and d 6.93 ppm. Electron Impact – Mass Spectrometry (EI-MS) of the compound exhibited M+ at m/z 330. Thus based on the Rf values, UV, 1H-NMR, 13C-NMR, and EIMS spectral studies, the structure of the compound has identified as 5, 4¢-Dihydroxy 6,8-dimethoxy 7-O-rhamnosyl flavones.
| Keywords: |
MPA, Rf values, UV, 1H-NMR, 13C-NMR, EIMS l
INTRODUCTION: Pisonia aculeata Linn. (Nyctaginaceae) is a large scandent shrub, which holds an important place in folklore medicine. It is extensively used by native medical practitioners and tribes for treating swelling, rheumatic pains, jaundice, and tumors.
Preliminary phytochemical screening of the extract showed the presence of alkaloids, triterpenes, phenolic compounds, flavonoids, and glycosides.
However, no studies to date have been able to demonstrate the isolation and characterization of active constituents. Pisonia aculeata was fractionated using benzene, diethyl ether, and ethyl acetate. Ethyl acetate fraction on concentration yielded a yellow solid, which was non-homogenous in TLC. Hence subjected to separation and purification on column chromatography in that silica gel column (60-120 mesh, 300 gm, 100 ´ 5 cm) solvents of increasing polarity. Fractions were collected & homogeneity was examined on TLC. A Fraction 77-90 on concentration yielded a pure yellowish homogeneous solid and was designated as compound I.
It gave dark green coloration with neutral ferric chloride and violet coloration with Molish’s reagent. In order to find out the nature of the glycoside, compound I was subjected to acid hydrolysis. The basic flavonoid structure of compound I and position of attachment of hydroxyl and other substituents were conveniently studied by recording UV spectrophotometer (Shimadzu 1601) in MeOH as well as in various Shift reagents. And the l max values were determined.
The compound was crystallized from alcohol as a pale yellow amorphous powder. It showed intense UV maxima at 273 nm (band II) and 329 nm (band I), indicating the flavone nature of it. Band I underwent a significant bathochromic shift of +60 nm on addition of NaOMe which suggested the presence of free 4¢-OH group in ring B. Absence of characteristic bathochromic shift (5-20 nm) on addition of NaOAc suggested that C-7 was not free. The absence of characteristic bathochromic shift on addition of NaOAc/H3BO3 indicated the absence of O-dihydroxy substituent in ring B. A consistent bathochromic shift of band II (14 nm) with AlCl3/ HCl indicated the presence of hydroxyl substituent at C-5 along with oxygen at C-6 position.
The 1H-NMR spectrum of compound I was recorded using AMX 400 (400 MHz) spectrometer (Plate 1) using DMSO-d6 as the solvent, and a complete assignment of protons were determined. A 13C-NMR spectrum of compound I was recorded using AMX 400 (100 MHz) spectrometer (Plate 2) using DMSO-d6 as the solvent, and the complete assignment of carbon was determined. In the 1H-NMR spectrum of the compound, I showed a pair of doublets in the aromatic region at d 7.89 ppm and d 6.93 ppm each integrating two protons indicated the presence of two A2 B2 pattern due to protons a C-3¢, C-5¢and C-2¢, C-6¢ respectively of ring B of flavone. This was supported by the UV shift experiments and 13C-NMR values.
Violet colouration of the compound with Molisch’s reagent indicated the presence of glycoside moiety. The position of glycosylation at C-7 as indicated by UV studies was confirmed by the presence of anomeric proton signal displayed at 5.15 ppm [For C-3 anomeric proton appears at d 5.8 ppm]. Rhamnosyl nature of the sugar and its attachment to C-7 carbon was confirmed by acid hydrolysis and 1H-NMR studies. The 1H-NMR spectrum also showed one singlet at d 6.2 ppm corresponding to C-3 proton of the flavone skeleton, which is also supported by the 13C-NMR signals at d 164. 9 (C-2), 102.3 (C-3), and a quaternary signal at 180.9 (C-4). The absence of other characteristic signals in the aromatic region of the 1H-NMR spectrum suggested that all the carbon atoms of ring A are substituted.
The 5-hydroxy and C-6, C-7, and C-8 substitution of ring A is further supported by the 13C-NMR values at d 157.6 (C-5), 161.1(C-7), 131.5(C-6), and 128.6 ppm (C-8). The absence of signals for H-6 and H-8 in 1H-NMR, the downfield shift of C-6 and C-8 in 13C-NMR4 and the appearance of two methoxyl signals at d 60.1 and d 56.1 ppm suggested the possibility of substitution of C-6 and C-8 by methoxyl groups.
Electron Impact – Mass Spectrometry (EI-MS) of the compound exhibited M+ at m/z 330. Fragment ion at m/z 212 and m/z 118 consistent with retero-Diel’s Alder fragmentation. Fragment ion at m/z 118 confirmed the presence of C-4¢ hydroxyl group in ring B. Thus based on the Rf values, UV, 1H-NMR, 13C-NMR, and EIMS spectral studies, the structure of compound has identified as 5, 4¢-Dihydroxy 6,8-dimethoxy 7-O-rhamnosyl flavones.
MATERIALS AND METHODS:
Isolation of Compound: The concentrate of the alcohol extract of Pisonia aculeata was fractionated using benzene (3 × 300 ml), diethyl ether (3 × 300 ml), and ethyl acetate (4 × 300 ml). The ethyl acetate fraction on concentration yielded a yellow solid, which was non-homogenous in TLC and hence was further subjected to separation and purification on column chromatography.
Column Chromatographic Analysis: The residue obtained from the ethyl acetate fraction (15 g) of Pisonia aculeata was chromatographed in silica gel column (60-120 mesh, 300 gm, 100 × 5 cm) using gradient elution with the solvents of increasing polarity. Fractions of 100 ml were collected each time, and the homogeneity was examined on TLC with suitable solvents. The details of the fractionations and their characteristics are given in Table 1. Fractions 1-5, 6-10, 11-48, 49-64 and 65-76 each of which on concentration yielded residues with varying intensities of yellow colour. These were tested individually by TLC, and further purification was not carried out because of the paucity of the samples. Fractions 77-90 on concentration yielded a pure yellowish homogeneous solid and was designated as compound I. It gave dark green colouration with neutral ferric chloride and violet colouration with Molish’s reagent. The Rf values of compound I in various solvent systems are given in Table 2.
Acid Hydrolysis of Compound I: In order to find out the nature of the glycoside, compound I was subjected to acid hydrolysis. To a solution of the glycoside (10 mg) in hot methanol (10 ml), an equal volume of H2SO4 (7%) was added, and the mixture was gently refluxed at 100 ºC for 2 h. The excess of alcohol was distilled off in vacuo, and the resulting aqueous solution was partitioned with ether to separate the ether soluble aglycone and the aqueous sugar.
Identification of the Sugar: The aqueous layer was treated with BaCO3 to remove excess sulphuric acid, and the barium sulphate formed was filtered off using Whatman no. 42 filter paper, and the filtrate (sugar portion) was concentrated. The concentrate was analysed by Paper chromatography (PC) with various authentic sugar samples on a Whatman no.1 filter paper strip and identified using Aniline hydrogen phthalate spray reagent (prepared by dissolving 9.2 ml of aniline and 16 g of phthalic acid in 490 ml of n-butanol, 490 ml of ether and 20 ml of water). The various solvent systems used for PC and Rf values of the identified sugar in these solvent systems are presented in Table 3.
UV Spectral Characteristics of Compound I: Basic flavonoid structure of compound I and position of attachment of hydroxy and other substituents were conveniently studied by recording UV spectrophotometer (Shimadzu 1601) in MeOH as well as in various Shift reagents. The λ max values are given in Table 4.
1H-NMR Spectral data of compound I: The 1H-NMR spectrum of compound I was recorded using AMX 400 (400 MHz) spectrometer (Plate 1) using DMSO-d6 as the solvent and complete assignment of protons are shown in Table 5.
13C-NMR Spectral Data of Compound I: 13C-NMR spectrum of compound I was recorded using AMX 400 (100 MHz) spectrometer (Plate 2) using DMSO-d6 as the solvent, and the complete assignment of carbon are given in Table 6.
EI-MS Study of Compound I: EI-MS spectrum (Fenniganmat 8230, 70 eV) of compound I (Plate 3) was taken, and it gave various fragments at m/z: 330, 213, and 118. The compound was crystallized from alcohol as a pale yellow amorphous powder. It showed intense UV maxima at 273 nm (band II) and 329 nm (band I), indicating the flavone nature of it. Band I underwent a significant bathochromic shift of + 60 nm on addition of NaOMe which suggested the presence of free 4¢-OH group in ring B. Absence of characteristic bathochromic shift (5-20 nm) on addition of NaOAc suggested that C-7 was not free.
The absence of characteristic bathochromic shift on the addition of NaOAc/H3BO3 indicated the absence of O-dihydroxy substituent in ring B. A consistent bathochromic shift of band II (14 nm) with AlCl3/ HCl indicated the presence of hydroxyl substituent at C-5 along with oxygen at the C-6 position (Mabry TJ, et al., 1970). In the 1H-NMR spectrum of a compound, I showed a pair of doublets in the aromatic region at d 7.89 ppm and d 6.93 ppm each integrating two protons indicated the presence of two A2 B2 pattern due to protons a C-3¢, C-5¢ and C-2¢, C-6¢ respectively of ring B of flavone. This was supported by the UV shift experiments and 13C-NMR values (Faini FA, et al., 1982). Violet colouration of a compound with Molisch’s reagent indicated the presence of glycoside moiety. The position of glycosylation at C-7 as indicated by UV studies was confirmed by the presence of anomeric proton signal displayed at 5.15 ppm [For C-3 anomeric proton appears at d 5.8 ppm] (Faini FA, et al., 1982) Fig. 2. Rhamnosyl nature of the sugar and its attachment to C-7 carbon was confirmed by acid hydrolysis and 1H-NMR studies.
The 1H-NMR spectrum also showed one singlet at d 6.2 ppm corresponding to C-3 proton of the flavone skeleton, which is also supported by the 13C-NMR signals at d 164.9 (C-2), 102.3 (C-3), and a quaternary signal at 180.9 (C-4) (Agarwal PK, 1989). The absence of other characteristic signals in the aromatic region of the 1H-NMR spectrum suggested that all the carbon atoms of ring A are substituted. The 5-hydroxy and C-6, C-7 and C-8 substitution of ring A is further supported by the 13C-NMR values at d 157.6 (C-5), 161.1(C-7), 131.5(C-6) and 128.6 ppm (C-8) (Agarwal PK, 1989). The absence of signals for H-6 and H-8 in 1H-NMR, the downfield shift of C-6 and C-8 in 13C-NMR4 and the appearance of two methoxyl signals at d 60.1 and d 56.1 ppm suggested the possibility of substitution of C-6 and C-8 by methoxyl groups.
EI-MS of the compound exhibited M+ m/z 330 and fragment ion at m/z 212 and m/z 118 consistent with retero-Diel’s Alder fragmentation, and a fragment ion at m/z 118 confirmed the presence of C-4¢ hydroxyl group in ring B. Thus based on the Rf values, UV, 1H-NMR, 13C-NMR, and EIMS spectral studies, the structure of the compound has identified as 5, 4¢-dihydroxy 6, 8-dimethoxy flavone skeleton Table 1-6, Fig. 1-4.
RESULTS:
TABLE 1: CHROMATOGRAPHIC FRACTIONS OF ETHYL ACETATE CONCENTRATE OF MPA
| Fractions Collected | % Eluent Composition | Remarks |
| 1-5
6-10 11-48 49-64 65-76 77-90 91-104 |
100 Petroleum ether
90/10 Pet. Ether/benzene 80/20 to 10/90 Pet. Ether/benzene 100 benzene 90/5 benzene /EtOAc 80/20 benzene /EtOAc 70/30 benzene /EtOAc |
Yellow waxy substance
Yellow waxy substance Pale yellow solid Pale yellow solid Yellow solid Yellow solid Yellow-brown solid |
TABLE 2: RF 100 VALUES OF COMPOUND IN PAPER CHROMATOGRAPHY
| Compound I |
Solvent System |
||||||
| 15% HOAc | 30% HOAc | 50% HOAc | 60%
HOAc |
BAW* | Forestal# | PhOH+ | |
| 11.26 | 31.34 | 78.76 | 83.60 | 20.83 | 93.84 | 34.42 | |
*BAW – n- butanol: acetic acid: water (4:1:5) m#Forestal- Acetic acid: Conc HCl: H2O (30:3:10) +PhOH- Phenol saturated with water (3:1)
TABLE 3: RF 100 VALUES OF SUGAR OF COMPOUND
| Sugar | Developing Solvents | |||
| BAW* | PhOH+ | Forestal# | EtOAc: Pyridine: H2O 10:4:3 | |
| Sugar from compound I | 38 | 55 | 58 | 54 |
| Authentic Rhamnose | 37 | 55 | 59 | 55 |
* BAW – n- butanol: acetic acid: water (4:1:5) + PhOH- Phenol saturated with water (3:1) #Forestal- Acetic acid: Conc HCl: H2O (30:3:10)
TABLE 4: LMAX VALUES OF COMPOUND
| Solvent / Shift Reagents | lmax (nm) |
| MeOH
+NaOMe +NaOAc +NaOAc +H3BO3 +AlCl3 +AlCl3 + HCl |
273, 329
273, 340, 389 273, 313 272, 329 270, 348 270, 300, 315, 347 |
TABLE 5: 1H-NMR DATA OF COMPOUND IN DMSO-D6
| d H (ppm) |
Signal Assignment |
| 12.95
7.89 6.93 6.38 5.15 3.9 3.8 3.0 - 3.75 1.2 |
1 H, s, 5-OH
2H, d, (J=8.7 Hz ), H-2¢ 6¢ 2H, d, (J=8.7 Hz), H-3¢ 5¢ 1H, s, H-3 1H, d, (J=2 Hz), H-1 of rhamnose 3H, s, OCH3 group 3H, s, OCH3 group Sugar protons 3H, d, J=6 Hz, H-6² |
TABLE 6: 13C-NMR DATA OF COMPOUND IN DMSO - D6
| C | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 | C-8 | C-9 | C-10 |
| Ppm | 164.9 | 102.3 | 180.9 | 157.6 | 131.5 | 161.1 | 128.6 | 148.0 | 105.5 |
| C | C-1¢ | C-2¢ | C-3¢ | C-4¢ | C-5¢ | C-6¢ |
| Ppm | 120.0 | 127.9 | 115.9 | 160.5 | 115.9 | 127.9 |
| C | C-1² | C-2² | C-3² | C-4² | C-5² | C-6² | OCH3 | OCH3 |
| Ppm | 98.4 | 70.1 | 69.9 | 71.8 | 69.9 | 18.7 | 60.1 | 56.1 |
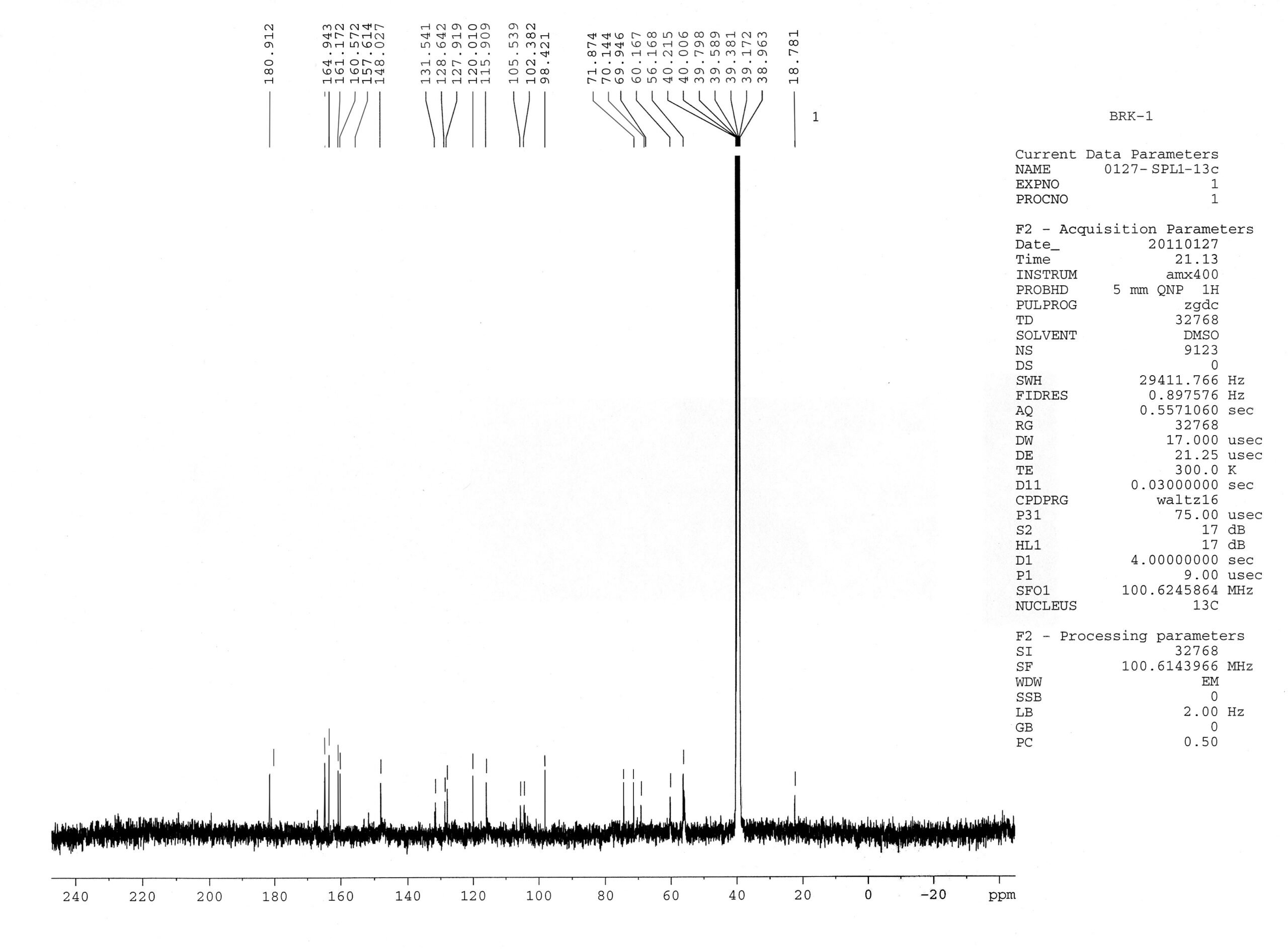
FIG. 1: BRK C1
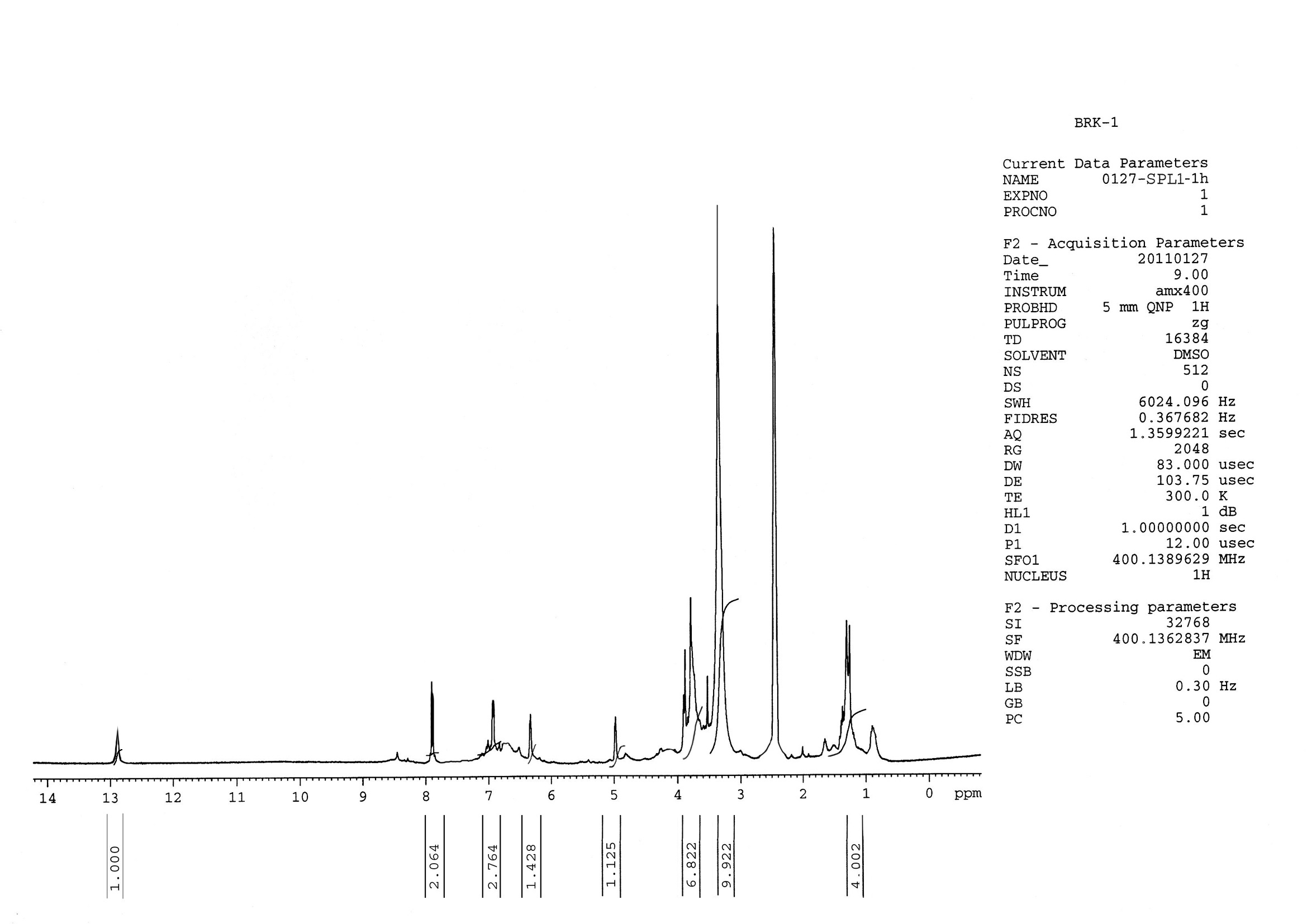
FIG. 2: BRK P1
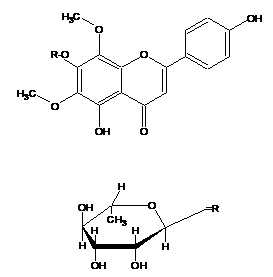
FIG. 3: 5, 4¢-DIHYDROXY 6, 8-DIMETHOXY 7-O-RHAMNOSYL FLAVONE
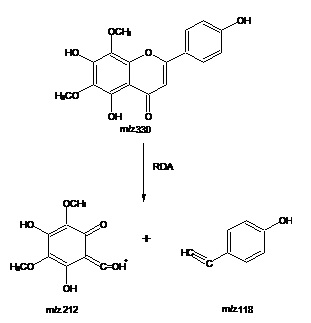
FIG. 4: EI-MS FRAGMENTATION
DISCUSSION: The focus of this paper is on the analysis of bioactive compounds present in the Pisonea aculeata Linn. Having anticancer activity involving the applications of chromatographic techniques such as TLC and column chromatography, Spectrophotometric techniques such as UV, 1H-NMR, 13C-NMR and EIMS spectral studies. The present investigation was carried out by fractionating the Methanolic extract of Pisonia aculeata (MPA) using benzene, diethyl ether, and ethyl acetate.
Ethyl acetate fraction on concentration subjected to separation and purification on column chromato-graphy. Fractions were collected & homogeneity was examined on TLC. A Fraction 77-90 on concentration yielded a pure yellowish homogeneous solid and gave dark green coloration with neutral ferric chloride and violet coloration with Molish’s reagent. In order to find out the nature of the glycoside, compound I was subjected to acid hydrolysis. The basic flavonoid structure of compound studied by recording UV spectro-photometer showed an intense UV maxima at 273 nm (band II) and 329 nm (band I), indicating the flavone nature of it. The 1H-NMR spectrum of compound I was recorded using AMX 400 (400 MHz) spectrometer using DMSO-d6 as the solvent showed a pair of doublets in the aromatic region at d 7.89 ppm and d 6.93 ppm. Electron Impact – Mass Spectrometry (EI-MS) of the compound exhibited M+ at m/z 330.
CONCLUSION: In the present study, based on the Rf values, UV, 1H-NMR, 13C-NMR, and EIMS spectral studies, the structure of the compound has been identified as 5, 4¢-Dihydroxy 6, 8-dimethoxy 7-O-rhamnosyl flavones from leaves of P. aculeate. The fact that these are the active compounds responsible for anticancer activity is evident from previous cytotoxic studies of the extract. These isolated compounds could also be active as an anticancer drug by studying the different human cancer cell lines.
ACKNOWLEDGEMENT: Nil
CONFLICTS OF INTEREST: Nil
REFERENCES:
- Agarwal PK: 13CNMR of flavonoids. Elsevier Amsterdam 1989; 15: 289.
- Anand PA, Kunnumakkara C, Sundaram K, Harikumar S, Tharakan, Lai O, Sung B and Aggarwal B:. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res 2008; September 25(9): 2097-16.
- Anonymous: The wealth of India - raw materials. Council for Scientific and Industrial Res New Delhi 2003; 8: 119.
- Archer MC: Mechanisms of action of N-nitroso compounds. Cancer Surv 1989; 8(2): 241-50.
- Australian Tropical Rainforest Plants, factsheet “Pisonea aculeata”, Family, Nyctaginaceae.
- Basset J, J Denny, J Jeffery and J: Mendham vogel's text book of quantitative inorganic analysis, 4th edition. ELBS-Longman Essex UK 1985; 196.
- Bersier P, J Bersier and M Smyth Eds Vos, JG: Analytical voltammetry. Elsevier Amsterdam 1992; 159.
- Burton K: A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Bio Chem J 1956; 62: 315-23.
- Dryhurst G and Niki K: Redox chemistry and interfacial behavior of biological molecules. Plenum Press New York 1988; 369.
- Gooding JJ: Electrochemical DNA hybridization biosensors. Electroanalysis 2002; 14: 1149-56.
- Henry RJ: Clinical chemistry principles and techniques, 2nd ed. Harper and Row Publication New York 1974; 225.
- Jasim H, Al-Temimi AA and Al-Amiery AH: Study the anticancer activities of ethanolic Curcumin extract. African Journal of Pure and Applied Chemistry 2010; 4(5): 68-73.
- Katzung BG: Basic and clinical pharmacology. in: cancer chemotherapy, (eds. sydney et al. Appleton & Lange Connecticut 1992; 766-67.
- Kauffmann J and J Vire: Pharmaceutical and biomedical application of electroanalysis: a critical review. Anal Chim Acta 1993; 173- 29.
- Kaufman PJ, Leland C, Warber A, James D, Harry and Brielmann L: Natural products from plants CRC Press London 1999; 1581-89.
How to cite this article:
Ghode SP, Ghode PD, Chatur VM and Kolhe R: Isolation and characterization of active constituents from plant Pisonia aculeata linn by spectral analysis. Int J Pharmacognosy 2021; 8(2): 82-88. doi link: http://dx.doi.org/10.13040/IJPSR.0975-8232.IJP.8(2).82-88.
This Journal licensed under a Creative Commons Attribution-Non-commercial-Share Alike 3.0 Unported License.
Article Information
8
82-88
666
903
English
IJP
S. P. Ghode *, P. D. Ghode, V. M. Chatur and R. Kolhe
Department of Pharmacognosy, Rasiklal M. Dhariwal Institute of Pharmaceutical Education and Research, Chinchwad, Pune, Maharashtra, India.
chintalwarshweta@gmail.com
15 December 2020
20 February 2021
25 February 2021
10.13040/IJPSR.0975-8232.IJP.8(2).82-88
28 February 2021