


IN SILICO PREDICTION OF RIBOSWITCH AS A POTENTIAL DRUG TARGET AND DESIGN OF ITS OPTIMAL INHIBITORS AND PHARMACOPHORE FOR BACILLUS CEREUS
HTML Full TextIN-SILICO PREDICTION OF RIBOSWITCH AS A POTENTIAL DRUG TARGET AND DESIGN OF ITS OPTIMAL INHIBITORS AND PHARMACOPHORE FOR BACILLUS CEREUS
Shant Swarup Jaiswal
Department of Mathematics and Bioinformatics, MANIT Bhopal - 462003, Madhya Pradesh, India.
ABSTRACT: Bacillus cereus is a gram-positive, rod-shaped, motile, beta hemolytic bacteria. These bacteria cause foodborne illness, severe nausea, vomiting, diarrhea, and keratitis. Foodborne illness caused by Bacillus cereus occurs because of the bacterial endospores survival in improperly cooked food. The bacteria produce enterotoxins having high resistance against heat and acids, and they cause the diarrheal and emetic syndrome. The diarrhetic syndromes observed in patients are thought to stem from the three toxins: hemolysin BL (Hbl), non-hemolytic enterotoxin (Nhe) and cytotoxin K (CytK). These enterotoxins are all produced in the small intestine of the host, thus thwarting digestion by endogenous host enzymes. The effect is the loss of cellular membrane potential and eventually cell death. Antibiotics that are used for the treatment of infectious disease have suffered a major setback due to the development of drug resistance by the pathogenic target organism. Therefore, in the current study, the riboswitches are explored as an alternative potential drug target. A riboswitch of Bacillus cereus bacteria has been identified as a drug target, and an inhibitor is designed with the help of molecular docking and simulation based virtual screening. Based on the in-silico studies, the designed Lead 1 molecule is recommended as a potential inhibitor for riboswitch of Bacillus cereus. The proposed inhibitor is superior over the existing antibiotics as it has less side effects and remote chances of development of resistance by the pathogen.
| Keywords: |
Riboswitch, Drug design, Molecular docking simulations, Virtual screening, Bacillus cereus, Pharmacophore
INTRODUCTION: Bacillus cereus is a spore-forming bacterium that can be frequently isolated from soil and some food. It causes gastrointestinal diseases in our body. If B. cereus grows in food, it can cause two different types of foodborne illness in humans vomiting very shortly after eating contaminated food or diarrhea after a longer incubation.
Two different toxins are responsible for that emetic toxin and the diarrheal toxin. It is the action of the toxin in the small intestine that causes diarrhea. In a small percentage of cases, both vomiting and diarrheal symptoms can occur if both types of toxins are produced. Spores of B. cereus can be found widely in nature, including samples of dust, dirt, cereal crops, water, etc., so it is a common contaminant of raw agricultural commodities.
The diarrhea-causing strains have been found in a wider selection of foods. Common sources include meat and vegetable items, soups and milk products. Unlike the emetic toxin, the diarrheal toxin is destroyed in cooking.
If the spores experience conditions that permit growth, they can grow to levels where toxins are produced. An additional concern regarding B. cereus is the resistance of the spores to per acetic acid treatments that may be used as an alternative to hydrogen peroxide for treatment of aseptic packaging materials. B. cereus spores are more resistant to some per acid products than other spore formers. Heating can activate spores, which allows them to germinate and grow under favorable conditions. Time and temperature abuse of cooked food permit the activated spores to grow and produce toxins.
Many of the emetic illnesses are due to improper holding of cooked rice at warm room temperatures, offering conditions where the activated spores in the rice can produce toxin.
Heating of a food after potential temperature abuse is not a foolproof control technique for B. cereus since the emetic toxin is heat stable. The diarrheal toxin will, however, be destroyed by heating. New validated cellular targets are needed to reinvigorate antibacterial drug discovery. This need could potentially be filled by riboswitches - messenger RNA (mRNA) structures that regulate gene expression in bacteria. Riboswitches are unique among RNAs that serve as drug targets in that they have evolved to form structured and highly selective receptors for small drug-like metabolites.
In most cases, metabolite binding to the receptor represses the expression of the gene(s) encoded by the mRNA. If a new metabolite analog were designed that binds to the receptor, the gene(s) regulated by that riboswitch could be repressed, with a potentially lethal effect to the bacteria.
Recent work suggests that certain antibacterial compounds discovered decades ago function at least in part by targeting riboswitches.
MATERIAL AND METHODS: The proposed framework for prediction and identification of riboswitches, their ligands, and design of their optimal inhibitors and Pharmacophore is given below.
Preparation of Input Files: Genes of Bacillus cereus are taken from NCBI in FASTA format. This data forms the input of the present study 1.
Identification of Gene Sequence for Riboswitch: Genes are searched for riboswitch like sequence through the use of online riboswitch identifying tools. Riboswitch finder and Rfam database are employed for the identification of riboswitches. Based on the sequence conservation, Riboswitch finder can detect specific sequence elements, and along with that, it’s also capable of searching 10 types of riboswitches against an input sequence. Whereas Rfam database is a collection of RNA sequence families and covariance models (CMs), and when it’s searching for riboswitches, it incorporated covariance models (CMs) and scored based on a combination of the consensus whole sequence and the consensus RNA ligand secondary structure 2, 3.
Prediction of Tertiary Structure of Riboswitches: The secondary and tertiary structure of riboswitch is predicted by using online iFold RNA tool. iFold RNA program is a web-based methodology for rapidly exploring RNA conformations using discrete molecular dynamics simulations (Sharma et al., 2008). The 3D structure of the riboswitch is further visualized using the GUI interface of the Chimera molecular modeling suite. Chimera is a protractile program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles (Pettersen et al., 2004 4).
Molecular Docking Simulation:
Two Types of Docking are Performed:
Blind Docking (BD): Blind docking is done by using the GUI interface of AutoDock. AutoDock software is downloaded from the Scripps portal (Morris et al., 2009). In blind docking; the appropriate binding site is searched (Shaikh et al., 2015) in the predicted riboswitch of B. Cereus. In the blind docking method, the whole riboswitch structure is covered under 3D grid box for docking. The coordinates of the grid box utilized for finding a binding site for the riboswitch 5.
Focused Docking (FD) for Identification of Ligand: For targeting the specific ligand binding site, focused grid box covering ligand as well as binding residues is prepared. Focused docking is performed by using 20 different amino acids and 11 different cofactor or metabolites which are involved in direct binding at 5’UTR of different riboswitches 6.
Refinement of Focus Docking Results: After identification of an appropriate ligand from focused docking, repeat the docking of the identified ligand with the riboswitch number of times to refine the results.
Virtual Screening Using NCI Diversity Set: Virtual screening of the NCI diversity set containing 1880 diverse molecules is done by using identified the binding site predicted in the structure of the riboswitch. The files required for virtual screening are prepared through Raccoon software. Raccoon (Stefano Forli, Online) is a graphical interface for processing ligand libraries in different formats (PDB, PDBQT) and to generate all files required to run an AutoDock virtual screening [Walters et al., 1998]. The coordinates of the grid box used in virtual screening of the riboswitch are same as docking and grid parameters used in focused docking 7.
Score and Toxicity of Lead Molecules Prediction: The top fourteen screened lead molecules are checked for toxicity and drug score with the help of OSIRIS online tool, in which seven lead molecules are passes the given parameters, and other seven do not pass the given parameters. Now we only consider these seven lead molecules and use OSIRIS to calculate various drug-relevant properties like cLogP value, molecular weight, Drug likeness, drug score and toxicities like mutagenicity, tumorigenicity, irritant effects and reproductive effects in lead molecules based on the functional group present in the structure of lead molecules. The results predicted in OSIRIS are color coded. Red color shows the properties with high risks of undesired effects like mutagenicity or poor intestinal absorption, whereas a green color indicates drug-conform behavior. The drug score combines a drug-likeness, cLogP, logS, molecular weight, and toxicity risk, which is used to judge the lead molecule overall potential to qualify for a drug (OSIRIS Property Explorer, Online).
Evaluation of Physicochemical Properties of Lead Compounds: After evaluating the toxicity of top seven screened lead molecules, their physiochemical properties like 2D Polar surface area (2D PSA), Partition Coefficient (cLogP), Molecular Weight, Hydrogen Donor, and Acceptor sites are checked using Marvin Sketch software (Marvin Sketch, Online).
Prediction of Pharmacophore: The top seven screened lead molecules are chosen, and with the use of PharmaGist online tool, their pharmacophore is identified. Pharmacophore is spatial arrangements of features that are essential for the molecule to interact with the specific target receptor and the PharmaGist is ligand based freely available web server for pharmacophore detection. The output of pharmacophore provides a 3D superposition of conformations of input ligands that share it (Duhovny et al., 2008) and this superposition can be easily analyzed by using Discovery Studio 3.5 downloaded from the accelrys website (Accelrys Software, Online 8).
Lead Modification: We can do bioisosteric substitution in pharmacophore. Bioisosteric compounds are those having similar valency. We are doing lead modification to increase its potency and decrease toxic effects if any 9.
Molecular Docking Simulation of Designed Pharmacophore - Based Lead Molecules with Riboswitch: After modification in pharmacophore through bioisosteric substitution we designed four lead molecules, they are docked with the riboswitch for predicting binding energy. The coordinates of the grid box used in docking are the same as used for focused docking 5, 6.
Evaluating Physiochemical Properties of Designed Pharmacophore-Based Lead Molecules: After docking of designed four lead molecules, evaluate their physiochemical properties like 2D Polar surface area (2D PSA), Partition Coefficient (cLogP), Molecular Weight, Hydrogen Donor, and Acceptor sites are checked using Marvin Sketch software (Marvin Sketch, Online) 11, 12.
Evaluating Drug Score and Toxicity of Designed Pharmacophore-Based Lead Molecules: The pharmacophores - based lead molecules are evaluated for toxicity and drug-likeness score with the help of OSIRIS tool 10.
Proposed Pharmacophore - Based Lead Molecules for Bacillus cereus: After evaluation of physiochemical properties and Lipinski's filter rule of five, toxicity studies Absorption, distribution, metabolism, excretion and toxicity (ADMET) studies of designed pharmacophore based lead molecules we proposed best lead molecules which inhibit the activity of B. cereus bacteria, and these molecules are superior to existing antibiotics molecules as no toxic effects were predicted and has remote chances of development of drug resistance by the bacteria against this molecule.
RESULTS:
Preparation of Input Files: Genes of Bacillus cereus (accession no. NC_004722.1) are taken from NCBI in FASTA format. This data forms the input of the present study.
Identification of Gene Sequence for Riboswitch: Riboswitch finder and Rfam database are employed for the identification of riboswitches. Only one riboswitch are found in the genome of B. cereu, and its gene sequence is:
ACGAUCCUUCAUAUAUCCUCAAAGAUAUGGUUUGAGAGUCUCUACCAGGUUACCGUAAACAACCUGACUAUGAAGGCAGU
Prediction of Tertiary Structure of Riboswitches: The secondary and tertiary structure of riboswitch is predicted by using an online iFoldRNA tool from the identified sequence of Riboswitch like elements present in the genome of Bacillus cereus bacteria and visualized using chimera tool.
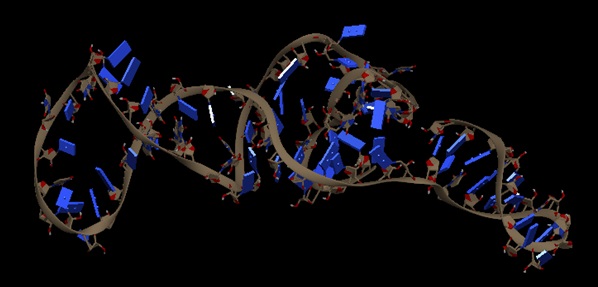
FIG. 1: TERTIARY STRUCTURE OF B. CEREUS RIBOSWITCH
Molecular Docking Simulation: Two types of docking are performed.
Blind Docking (BD): Blind docking is performed by using the GUI interface of AutoDock downloaded from the scripps portal. The suitable binding site is searched in the predicted riboswitch of Bacillus cereus using blind docking. In blind docking whole structure of riboswitch is covered under 3D grid box for docking. The coordinates of the grid box of riboswitches are given below.
TABLE 1: THE COORDINATES OF GRID BOX USED IN BLIND DOCKING
| Riboswitch | BCERS |
| x-D | 58 |
| y-D | 60 |
| z-D | 60 |
| Spacing (Angstrom) | 0.753 |
| X center | 6.445 |
| Y center | 1.554 |
| Z center | 14.29 |
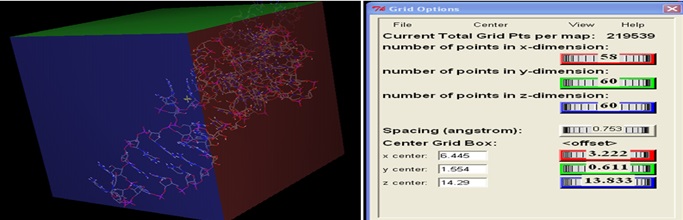
FIG. 2: GRID BOX USED FOR BLIND DOCKING OF CEREUS RIBOSWITCH COVERING the WHOLE MOLECULE INVOLVE IN BINDING OF LIGAND
As Alanine is neutral and simplest amino acids among all the 20 amino acids; therefore it is used as a ligand to predict the binding site in the riboswitch during blind docking. Appropriate binding sites are identified based on lowest binding energy in the range of -5 to -15 Kcal / Mol. The identified binding residue present in the riboswitch is further utilized for focused docking. The whole structure of Cereus Riboswitch and Alanine is covered by the grid using coordinate values as given in Table 1. Alanine will bind to a particular binding site and gives some energy shown in Table 2.
TABLE 2: BLIND DOCKING RESULT OF CEREUS RIBOSWITCH
| Riboswitch | Cereus |
| Ligand | Alanine |
| Binding energy (Kcal/Mol) | -4.43 |
| Ki (uM) | 566.69 |

FIG. 3: BINDING CONFORMATION OF THE LIGAND WITHIN THE ACTIVE BINDING BINDING SITE OF THE CEREUS RIBOSWITCH
Focused Docking (FD) for Identification of Ligand: For focusing the specific ligand binding site, focused grid box covering ligand as well as prepared binding residue. 20 different amino acid and 11 different cofactor or metabolites are using to perform focused docking which is involved in direct binding at 5 UTR of different riboswitches. The coordinates of the grid box used in focused docking of the riboswitch are given below.
TABLE 3: THE COORDINATES OF GRID BOX USED IN FOCUSED DOCKING
| Riboswitch | BCERS |
| x-D | 48 |
| y-D | 50 |
| z-D | 50 |
| Spacing(Angstrom) | 0.681 |
| X center | 3.028 |
| Y center | 1.554 |
| Z center | 13.39 |
After identification of an appropriate ligand from focused docking, repeat the docking of the identified ligand with the riboswitch number of times to refine the results. Obtained binding sites from blind docking is again covered by the grid box coordinates given in Table 3, and docked with 20 amino acids and 11 metabolites which have an affinity of binding on aptamer site of different Riboswitches and are checked for lowest binding energy as shown in tabulated form in Table 4 and Fourteen screened lead molecules with their respective binding energies shown in Table 5.
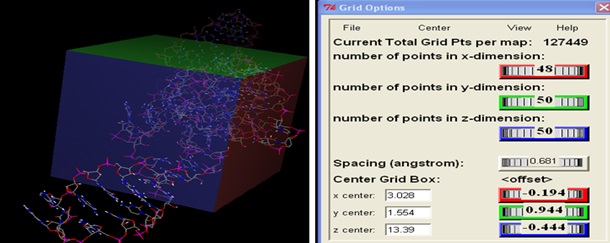
FIG. 4: GRID BOX USED FOR FOCUSED DOCKING OF CEREUS RIBOSWITCH COVERING ALL BINDING RESIDUES INVOLVE IN BINDING OF LIGAND
TABLE 4: RESULT OF FOCUSED DOCKING
| S. no. | Ligand | Binding Energy (Kcal/Mol) | Ki-value |
| 1 | Alanine | -4.43 | 566.69uM |
| 2 | Arginine | -6.89 | 8.91uM |
| 3 | Asparagine | -4.63 | 402.17uM |
| 4 | Aspartic acid | -3.05 | 5.83mM |
| 5 | Cysteine | -4.61 | 416.71uM |
| 6 | Glutamine | -4.94 | 241.15uM |
| 7 | Glutamic acid | -3.1 | 5.33mM |
| 8 | Glycine | -4.5 | 506.98uM |
| 9 | Histidine | -4.56 | 451.68uM |
| 10 | Isoleucine | -4.89 | 260.27uM |
| 11 | Leucine | -4.46 | 540.94uM |
| 12 | Lysine | -6.73 | 11.7uM |
| 13 | Methionine | -4.46 | 542.25uM |
| 14 | Phenylalanine | -5.67 | 69.78uM |
| 15 | Proline | -6.78 | 10.65uM |
| 16 | Serine | -4.67 | 377.25uM |
| 17 | Threonine | -4.76 | 325.81uM |
| 18 | Tryptophan | -5.46 | 99.63uM |
| 19 | Tyrosine | -5.03 | 205.31uM |
| 20 | Valine | -4.62 | 408.81uM |
| 21 | Adenine | -3.48 | 2.81mM |
| 22 | Cyclic diGMP | -8.13 | 1.09uM |
| 23 | Flavin mononucleotide | -3.19 | 4.63mM |
| 24 | Glucosamine-6-phosphate | -5.56 | 84.24uM |
| 25 | Guanine | -6.31 | 23.87uM |
| 26 | Queuosine | -10.52 | 19.5nM |
| 27 | S-adenosyl methionine | -4.68 | 368.19uM |
| 28 | S-adenosyl homocysteine | -5.72 | 64.15uM |
| 29 | Tertrahydro folate | -6.12 | 32.52uM |
| 30 | Thiamin pyrophosphate | -1.85 | 44.4mM |
Queuosine shows the minimum binding energy of -10.52 Kcal/Mol. Therefore, this ligand is chosen for further refinement process. The binding sites which we obtained from blind docking are further covered under the grid box by using the coordinates as given in Table 3 and docked with 20 amino acids and 10 metabolites which have an affinity of binding on aptamer site of different Riboswitches and are checked for lowest binding energy as shown in Table 4. Queuosine shows the minimum binding energy of -10.52 Kcal/Mol. Therefore, this ligand is chosen for further refinement process. Through virtual screening of NCI diversity set containing 1880 diverse molecules, many lead molecules are screened and are arranged according to a minimum to maximum binding energy 7. Among those lead molecules, fourteen lead molecules are selected randomly of different binding energies and are shown in Table 5.
TABLE 5: FOURTEEN SCREENED LEAD MOLECULES WITH THEIR RESPECTIVE BINDING ENERGIES
| Serial no. | ZINC ID | Structure | Binding energy |
| 1 | ZINC19325788_BCERS | 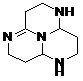 |
-16.73 |
| 2 | ZINC19325791_BCERS | 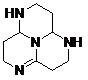 |
-15.65 |
| 3 | ZINC19325794_BCERS | 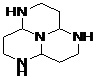 |
-15.36 |
| 4 | ZINC01673377_BCERS | 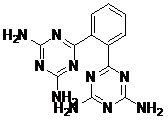 |
-14.11 |
| 5 | ZINC19230120_BCERS |  |
-11.82 |
| 6 | ZINC01556940_BCERS | 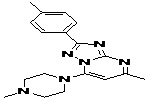 |
-11.38 |
| 7 | ZINC08652230_1_BCERS | 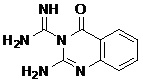 |
-10.75 |
| 8 | ZINC01584497_BCERS | 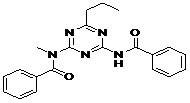 |
-10.74 |
| 9 | ZINC19362650_BCERS | 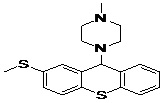 |
-10.66 |
| 10 | ZINC18163421_BCERS | 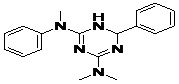 |
-10.39 |
| 11 | ZINC19366110_BCERS | 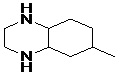 |
-10.27 |
| 12 | ZINC18189380_BCERS | 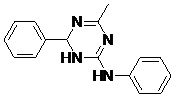 |
-10.12 |
| 13 | ZINC19362651_BCERS | 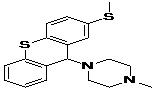 |
-10.07 |
| 14 | ZINC01669572_BCERS | 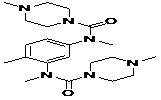 |
-10.04 |
Refinement of Focus Docking Results: After identification of an appropriate ligand from focused docking, repeat the docking of the identified ligand with the riboswitch number of times to refine the results.
Virtual Screening Using NCI Diversity Set: Virtual screening of the NCI diversity set containing 1880 diverse molecules is done by using identified the binding site predicted in the structure of the riboswitch. The files required for virtual screening are prepared through Raccoon software. Raccoon (Stefano Forli, Online) is a graphical interface for processing ligand libraries in different formats (PDB, PDBQT) and to generate all files required to run an AutoDock virtual screening (Walters et al., 1998 7). The coordinates of the grid box used in virtual screening of the riboswitch are same as docking and grid parameters used in focused docking.
Drug Score and Toxicity of Lead Molecules Prediction: We took the top fourteen lead molecules for finding the toxicity and drug score by TEST (Toxicity estimation software tool) tool. There are different drug-relevant properties are calculated by TEST tool like cLogP value, molecular weight, drug-likeness, drug score and toxicities like mutagenicity, tumorigenicity, irritant effects and reproductive effect in lead molecules based on the functional group present in lead molecules structure.
The toxicity and drug scores of all fourteen screened lead molecules predicted by using the OSIRIS online tool for Cereus riboswitch and select that lead molecule which shows no toxicity. Seven lead molecules are found to be no toxicity are shown in below table.
TABLE 6: TOXICITY AND DRUG SCORE PREDICTION OF SEVEN LEAD MOLECULES
| ID | Mutagenic | Tumorigenic | Irritant | Reproductive effect | Drug score |
| ZINC08652230_1 | LOW | LOW | LOW | LOW | 0.95 |
| ZINC19362650 | LOW | LOW | LOW | LOW | 0.71 |
| ZINC18163421 | LOW | LOW | LOW | LOW | 0.80 |
| ZINC19366110 | LOW | LOW | LOW | LOW | 0.59 |
| ZINC18189380 | LOW | LOW | LOW | LOW | 0.60 |
| ZINC19362651 | LOW | LOW | LOW | LOW | 0.71 |
| ZINC01669572 | LOW | LOW | LOW | LOW | 0.84 |
Evaluation of Physicochemical Properties of Lead Compounds: In physiochemical properties, the Lipinski’s rule of five criteria of seven lead molecules have been evaluated in Table 7.
TABLE 7: PHYSIOCHEMICAL PROPERTIES OF PROPOSED LEAD MOLECULES
| ID | ClogP | PSA | Mol. Wt | HBD | HBA |
| ZINC08652230_1 | -0.58 | 108.5 | 203 | 3 | 5 |
| ZINC19362650 | 4.31 | 57.08 | 342 | 0 | 2 |
| ZINC18163421 | 2.42 | 43.23 | 307 | 1 | 5 |
| ZINC19366110 | 0.75 | 24.06 | 154 | 2 | 2 |
| ZINC18189380 | 1.55 | 74.8 | 265 | 3 | 5 |
| ZINC19362651 | 4.31 | 57.08 | 342 | 0 | 2 |
| ZINC01669572 | 2.06 | 53.58 | 402 | 0 | 4 |
Prediction of Pharmacophore: Pharmacophore is designed by Pharma Gist online tool and visualized through discovery studio. The structures of pharmacophore are drawn by using Chem Draw.
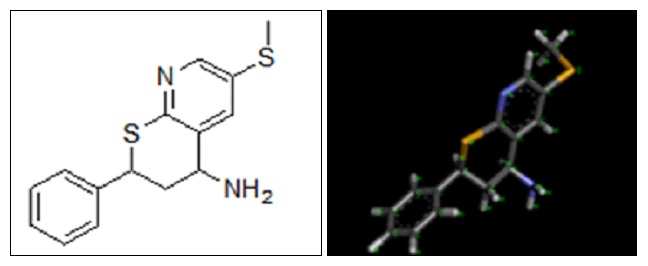
FIG. 5: 2D AND 3D STRUCTURE OF PREDICTED PHARMACOPHORE
Lead Modification: We can do bioisosteric substitution in pharmacophore. Bioisosteric compounds are those having similar valency. We are doing lead modification to increase its potency and decrease toxic effects if any.
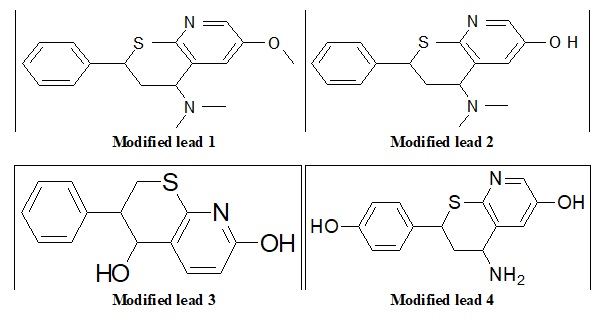
FIG. 6: STRUCTURE OF MODIFIED LEAD MOLECULES
Molecular Docking Simulation of Designed Pharmacophore - Based Lead Molecules with Riboswitch: We dock our modified lead molecules with cereus riboswitch receptor. For this we consider coordinates of focused docking, then make a table of its binding energy and KI - value.
TABLE 8: BINDING ENERGIES OF DESIGNED PHARMACOPHORE - BASED LEAD MOLECULES
| Pharmacophore | Structure | Binding energy | KI-Value (uM) |
| Lead 1 |  |
-7.41 | 3.694 |
| Lead 2 | 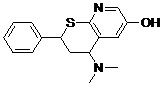 |
-7.22 | 5.11 |
| Lead 3 | 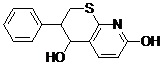 |
-5.01 | 211.33 |
| Lead 4 | 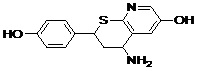 |
-7.04 | 6.94 |
Evaluating Physiochemical Properties of Designed Pharmacophore - Based Lead Molecules: In physiochemical properties, the Lipinski’s rule of five criteria of our modified lead molecules has been evaluated using OSIRIS online tool. Physicochemical properties such as 2D polar surface area (2D PSA), Partition Coefficient (cLogP), Molecular Weight, Hydrogen Bond Donor, and Acceptor sites are also checked using Marvin Sketch.
TABLE 9: PHYSIOCHEMICAL PROPERTIES OF DESIGNED PHARMACOPHORE-BASED LEAD MOLECULES
| Pharmacophore | clogP | PSA | Mol. weight | HBD | HBA |
| Lead 1 | 2.75 | 50.66 | 300 | 0 | 3 |
| Lead 2 | 2.48 | 61.66 | 286 | 1 | 3 |
| Lead 3 | 2.26 | 78.65 | 259 | 2 | 3 |
| Lead 4 | 1.15 | 104.6 | 274 | 3 | 4 |
Evaluating Drug Score and Toxicity of Designed Pharmacophore - Based Lead Molecules: The pharmacophores - based lead molecules are evaluated for toxicity and drug-likeness score with the help of OSIRIS tool.
TABLE 10: TOXICITY AND DRUG SCORE PREDICTION OF DESIGNED PHARMACOPHORE - BASED LEAD MOLECULES
| Pharmacophore | Mutagenic | Tumorigenic | Irritant | Reproductive effect | Drug score |
| Lead 1 | Low | Low | Low | Low | 0.88 |
| Lead 2 | Low | Low | Low | Low | 0.90 |
| Lead 3 | Low | Low | Low | Low | 0.89 |
| Lead 4 | Low | Low | Low | Low | 0.90 |
CONCLUSION: On the basis of predicted values, it can be concluded that four lead molecules lead 1, lead 2, lead 3, lead 4 can act as potential inhibitors against the Cereus Riboswitch of Staphylococcus species as all the four lead molecules have a good binding energy with the identified bacterial Riboswitch with optimum pharmacokinetics properties. All the four lead molecules are obtained by molecular docking simulation based virtual screening technique. These four lead molecules can inhibit the growth of Staphylococcus bacterial species by targeting their Cereus Riboswitch, and this Riboswitch sequence has not been reported in human beings.
Therefore, these designed lead molecules are not likely to interact with mammalian mRNA. Among all the four proposed lead molecules lead 1 molecule have high binding energy, so it is proposed to be the best optimal inhibitor molecule for riboswitch of Bacillus cereus, and it is also claimed that this molecule is superior to existing antibiotics molecules as no toxic effects were predicted and has remote chances of development of drug resistance by the bacteria against this molecule.
ACKNOWLEDGEMENT: Nil
CONFLICT OF INTEREST: Nil
REFERENCES:
- Dennis B, David JL and James O: “Genbank,” Nucleic Acids Research 1993; 21(13): 2963 -2965.
- Abreu-Goodger C and Merino E: RibEx: a web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic Acids Res 2005; 33: W690-2.
- Altschul SF, Gish W, Miller W, Myers EW and Lipman DJ: Basic local alignment search tool. J Mol Biol 1990; 5(3): 403-10.
- Sharma S, Feng D and Nikolay VD: "iFoldRNA: three-dimensional RNA structure prediction and folding". Advance Access Publication 2008; 24(17): 1951-1952.
- Dario G and Roberto S: Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins 2008; 74: 417-424.
- Bachwani M and Kumar R: "Molecular docking" IJRAP 2011; 2(6): 1746-1751.
- Sandro C, Stefano F, Alex LP, Rodney H, David SG and Arthur JO: Virtual Screening with Auto Dock: Theory and Practice. Expert Opin Drug Discov 2010; 5(6): 597-607. doi:10.1517/17460441.2010.484460.
- Inbar Y, Schneidman-Duhovny D, Dror O, Nussinov R and Wolfson HJ: Deterministic pharmacophore detection via multiple flexible alignment of drug-like molecules. In Proc. of RECOMB of Lecture Notes in Computer Science 2007; 3692: 423-434.
- Brown N: (Ed.) Bioisosteres in Medicinal Chemistry. Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany 2012.
- Nicolaou CA and Brown N: Multi-objective optimization methods in drug design. Drug Discovery Today: Technol. 2013; 10: e427-e435.
- Klinke DJ and Wang Q: Computational and Mathematical Methods in Medicine 2012, Article ID 676015.
- Neervannan S: Preclinical formulations for discovery and toxicology: physicochemical challenges. Expert Opinion on Drug Metabolism & Toxicology 2006; 2: 715-731.
How to cite this article:
Jaiswal SS: In silico prediction of riboswitch as a potential drug target and design of its optimal inhibitors and pharmacophore for Bacillus cereus. Int J Pharmacognosy 2018; 5(1): 26-36. doi link: http://dx.doi.org/10.13040/IJPSR.0975-8232.IJP.5(1).26-36.
This Journal licensed under a Creative Commons Attribution-Non-commercial-Share Alike 3.0 Unported License.